A Universal Framework for Accurate and Efficient Geometric Deep Learning of Molecular Systems
分子系统精确高效几何深度学习的通用框架
ABSTRACT
摘要
Molecular sciences address a wide range of problems involving molecules of different types and sizes and their complexes. Recently, geometric deep learning, especially Graph Neural Networks (GNNs), has shown promising performance in molecular science applications. However, most existing works often impose targeted inductive biases to a specific molecular system, and are inefficient when applied to macromolecules or large-scale tasks, thereby limiting their applications to many real-world problems. To address these challenges, we present PAMNet, a universal framework for accurately and efficiently learning the representations of three-dimensional (3D) molecules of varying sizes and types in any molecular system. Inspired by molecular mechanics, PAMNet induces a physics-informed bias to explicitly model local and non-local interactions and their combined effects. As a result, PAMNet can reduce expensive operations, making it time and memory efficient. In extensive benchmark studies, PAMNet outperforms state-of-the-art baselines regarding both accuracy and efficiency in three diverse learning tasks: small molecule properties, RNA 3D structures, and protein-ligand binding affinities. Our results highlight the potential for PAMNet in a broad range of molecular science applications.
分子科学研究涉及各种类型和尺寸的分子及其复合物的广泛问题。近年来,几何深度学习(尤其是图神经网络 (GNNs))在分子科学应用中展现出优异性能。然而,现有研究大多针对特定分子系统引入定向归纳偏置,在处理大分子或大规模任务时效率低下,限制了其在现实问题中的应用。为解决这些挑战,我们提出了PAMNet——一个通用框架,能够准确高效地学习任何分子系统中不同尺寸和类型的三维 (3D) 分子表征。受分子力学启发,PAMNet通过物理信息偏置显式建模局部与非局部相互作用及其协同效应,从而减少昂贵计算操作,实现时间和内存高效性。在涵盖小分子性质、RNA三维结构和蛋白质-配体结合亲和力的三大学习任务基准测试中,PAMNet在准确性与效率方面均超越现有最优基线。这些结果表明PAMNet在分子科学领域具有广泛的应用潜力。
Introduction
引言
The wide variety of molecular types and sizes poses numerous challenges in the computational modeling of molecular systems for drug discovery, structural biology, quantum chemistry, and others1. To address these challenges, recent advances in geometric deep learning (GDL) approaches have become increasingly important2,3. Especially, Graph Neural Networks (GNNs) have demonstrated superior performance among various GDL approaches 4–6. GNNs treat each molecule as a graph and perform message passing scheme on it7. By representing atoms or groups of atoms like functional groups as nodes, and chemical bonds or any pairwise interactions as edges, molecular graphs can naturally encode the structural information in molecules. In addition to this, GNNs can incorporate symmetry and achieve invariance or e qui variance to transformations such as rotations, translations, and reflections 8, which further contributes to their effectiveness in molecular science applications. To enhance their ability to capture molecular structures and increase the expressive power of their models, previous GNNs have utilized auxiliary information such as chemical properties 9–12, atomic pairwise distances in Euclidean space7,13,14, angular information 15–18, etc.
分子类型和尺寸的多样性给药物发现、结构生物学、量子化学等领域的分子系统计算建模带来了诸多挑战[1]。为解决这些挑战,几何深度学习 (geometric deep learning, GDL) 方法的最新进展显得愈发重要[2,3]。其中,图神经网络 (Graph Neural Networks, GNNs) 在各种GDL方法中展现出卓越性能[4-6]。GNNs将每个分子视为图结构,并在此基础上执行消息传递机制[7]。通过将原子或官能团等原子团表示为节点,化学键或任何成对相互作用表示为边,分子图能自然地编码分子结构信息。此外,GNNs还能整合对称性,实现对旋转、平移和反射等变换的不变性或等变性[8],这进一步提升了其在分子科学应用中的有效性。为增强分子结构捕捉能力并提高模型表达能力,现有GNNs已利用化学性质[9-12]、欧氏空间中的原子间距离[7,13,14]、角度信息[15-18]等辅助信息。
In spite of the success of GNNs, their application in molecular sciences is still in its early stages. One reason for this is that current GNNs often use targeted inductive bias for modeling a specific type of molecular system, and cannot be directly transferred to other contexts although all molecule structures and their interactions follow the same law of physics. For example, GNNs designed for modeling proteins may include operations that are specific to the structural characteristics of amino acids19,20, which are not relevant for other types of molecules. Additionally, GNNs that incorporate comprehensive geometric information can be computationally expensive, making them difficult to scale to tasks involving a large number of molecules (e.g., high-throughput compound screening) or macromolecules (e.g., proteins and RNAs). For instance, incorporating angular information can significantly improve the performance of $\mathrm{GNNs^{15-18}}$ , but also increases the complexity of the model, requiring at least $O(N k^{2})$ messages to be computed where $N$ and $k$ denote the number of nodes and the average degree in a graph.
尽管图神经网络 (GNN) 取得了成功,但它们在分子科学中的应用仍处于早期阶段。其中一个原因是当前的 GNN 通常针对特定类型的分子系统使用定向归纳偏置建模,无法直接迁移到其他场景——尽管所有分子结构及其相互作用都遵循相同的物理定律。例如,为蛋白质建模设计的 GNN 可能包含氨基酸结构特征特有的操作 [19,20],这些操作与其他类型分子无关。此外,整合全面几何信息的 GNN 计算成本高昂,难以扩展到涉及大量分子 (如高通量化合物筛选) 或大分子 (如蛋白质和 RNA) 的任务。例如,引入角度信息能显著提升 $\mathrm{GNN^{15-18}}$ 的性能,但也会增加模型复杂度,需要计算至少 $O(N k^{2})$ 条消息 (其中 $N$ 和 $k$ 分别表示图中的节点数和平均度数)。
To tackle the limitations mentioned above, we propose a universal GNN framework, Physics-Aware Multiplex Graph Neural Network (PAMNet), for the accurate and efficient representation learning of 3D molecules ranging from small molecules to macromolecules in any molecular system. PAMNet induces a physics-informed bias inspired by molecular mechanics 21, which separately models local and non-local interactions in molecules based on different geometric information. To achieve this, we represent each molecule as a two-layer multiplex graph, where one plex only contains local interactions, and the other plex contains additional non-local interactions. PAMNet takes the multiplex graphs as input and uses different operations to incorporate the geometric information for each type of interaction. This flexibility allows PAMNet to achieve efficiency by avoiding the use of computationally expensive operations on non-local interactions, which consist of the majority of interactions in a molecule. Additionally, a fusion module in PAMNet allows the contribution of each type of interaction to be learned and fused for the final feature or prediction. To preserve symmetry, PAMNet utilizes E(3)-invariant representations and operations when predicting scalar properties, and is extended to predict E(3)-e qui variant vectorial properties by considering the geometric vectors in molecular structures that arise from quantum mechanics.
为解决上述局限性,我们提出了一种通用图神经网络框架——物理感知多重图神经网络 (Physics-Aware Multiplex Graph Neural Network, PAMNet),用于准确高效地学习从有机小分子到任意分子系统中大分子的三维分子表征。PAMNet引入受分子力学[21]启发的物理先验偏置,基于不同几何信息分别建模分子中的局部与非局部相互作用。为此,我们将每个分子表示为双层多重图 (multiplex graph),其中一层仅包含局部相互作用,另一层则包含额外的非局部相互作用。PAMNet以多重图作为输入,针对不同类型的相互作用采用差异化操作整合几何信息。这种灵活性使PAMNet能通过避免对占分子相互作用主体的非局部作用进行昂贵计算来实现高效性。此外,PAMNet中的融合模块可学习各类相互作用的贡献度并融合为最终特征或预测结果。为保持对称性,PAMNet在预测标量性质时采用E(3)等变表征与运算,并通过考虑量子力学产生的分子结构几何向量,进一步扩展至预测E(3)等变矢量性质。
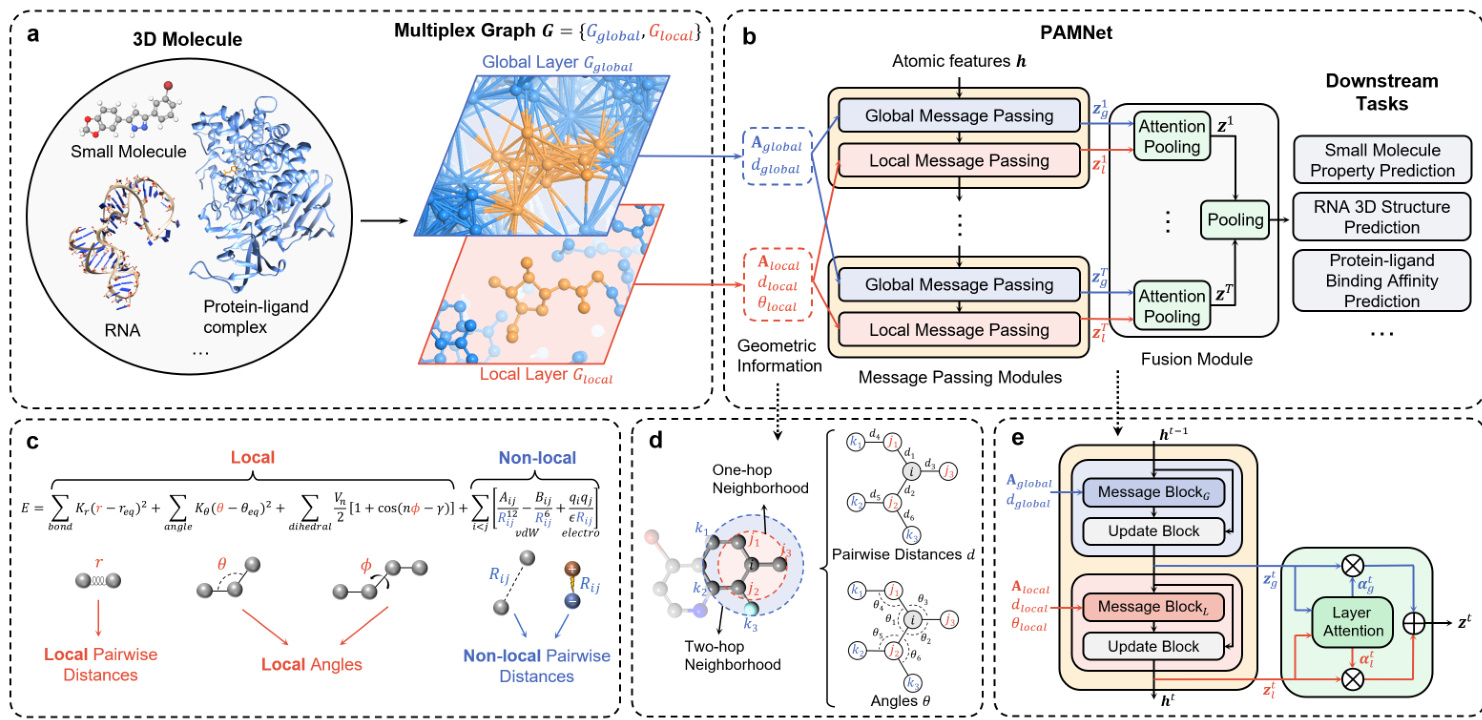
Figure 1. Overview of PAMNet. a, Based on the 3D structure of any molecule or molecular system, a two-layer multiplex graph $G={G_{g l o b a l},G_{l o c a l}}$ is constructed to separate the modeling of global and local interactions. b, PAMNet takes $G$ as input and learns node-level or graph-level representation for downstream tasks. PAMNet contains stacked message passing modules that update the node embeddings $\mathbf{z}$ in $G$ , and a fusion module that learns to combine the updated embeddings. In each message passing module, two message passing schemes are designed to encode the different geometric information in $G$ ’s two layers. In the fusion module, a two-step pooling process is proposed. c, Calculation of molecular energy $E$ in molecular mechanics. d, An example of the geometric information in $G$ . By considering the one-hop neighbors ${j}$ and two-hop neighbors ${k}$ of atom $i$ , we can define the pairwise distances $d$ and the related angles θ . e, Detailed architecture of the message passing module and the attention pooling in PAMNet.
图 1: PAMNet概述。a) 基于任意分子或分子系统的3D结构,构建双层复用图 $G={G_{g l o b a l},G_{l o c a l}}$ 以分离全局和局部相互作用的建模。b) PAMNet以 $G$ 作为输入,学习节点级或图级表征用于下游任务。PAMNet包含堆叠的消息传递模块(用于更新 $G$ 中的节点嵌入 $\mathbf{z}$ )和融合模块(学习组合更新后的嵌入)。每个消息传递模块中设计了两种消息传递方案,分别编码 $G$ 两层中的不同几何信息。融合模块提出了两步池化过程。c) 分子力学中分子能量 $E$ 的计算。d) $G$ 中几何信息的示例。通过考虑原子 $i$ 的一跳邻居 ${j}$ 和二跳邻居 ${k}$ ,可定义成对距离 $d$ 和相关角度θ。e) PAMNet中消息传递模块和注意力池化的详细架构。
To demonstrate the effectiveness of PAMNet, we conduct a comprehensive set of experiments on a variety of tasks involving different molecular systems, including small molecules, RNAs, and protein-ligand complexes. These tasks include predicting small molecule properties, RNA 3D structures, and protein-ligand binding affinities. We compare PAMNet to state-of-the-art baselines in each task and the results show that PAMNet outperforms the baselines in terms of both accuracy and efficiency across all three tasks. Given the diversity of the tasks and the types of molecules involved, the superior performance of PAMNet shows its versatility to be applied in various real-world scenarios.
为验证PAMNet的有效性,我们在涉及小分子、RNA和蛋白质-配体复合物等多种分子系统的任务上开展了一系列综合实验。这些任务包括预测小分子性质、RNA三维结构以及蛋白质-配体结合亲和力。我们将PAMNet与各任务中最先进的基线方法进行对比,结果表明PAMNet在所有三项任务的准确性和效率方面均优于基线方法。鉴于任务多样性和所涉分子类型的广泛性,PAMNet的卓越性能展现了其在多种现实场景中的应用潜力。
Overview of PAMNet
PAMNet 概述
Multiplex graph representation. Given any 3D molecule or molecular system, we define a multiplex graph representation as the input of our PAMNet model based on the original 3D structure (Fig. 1a). The construction of multiplex graphs is inspired by molecular mechanics 21, in which the molecular energy $E$ is separately modeled based on local and non-local interactions (Fig. 1c). In detail, the local terms $ E_{\mathrm{bond}}+E_{\mathrm{angle}}+E_{\mathrm{dihedral}}$ model local, covalent interactions including $E_{\mathrm{bond}}$ that depends on bond lengths, $E_{\mathrm{angle}}$ on bond angles, and $E_{\mathrm{dihedral}}$ on dihedral angles. The non-local terms $E_{\mathrm{vdW}}+E_{\mathrm{electro}}$ model non-local, non-covalent interactions including van der Waals and electrostatic interactions which depend on inter atomic distances. Motivated by this, we also decouple the modeling of these two types of interactions in PAMNet. For local interactions, we can define them either using chemical bonds or by finding the neighbors of each node within a relatively small cutoff distance, depending on the given task. For global interactions that contain both local and non-local ones, we define them by finding the neighbors of each node within a relatively large cutoff distance. For each type of interaction, we use a layer to represent all atoms as nodes and the interactions as edges. The resulting layers that share the same group of atoms form a two-layer multiplex graph $G={G_{g l o b a l},G_{l o c a l}}$ which represents the original 3D molecular structure (Fig. 1a).
多重图表示。给定任意3D分子或分子系统,我们基于原始3D结构定义了作为PAMNet模型输入的多重图表示 (图 1a)。多重图的构建灵感来源于分子力学[21],其中分子能量$E$根据局部与非局部相互作用分别建模 (图 1c)。具体而言,局部项$E_{\mathrm{bond}}+E_{\mathrm{angle}}+E_{\mathrm{dihedral}}$建模共价键局部相互作用,包括取决于键长的$E_{\mathrm{bond}}$、键角的$E_{\mathrm{angle}}$以及二面角的$E_{\mathrm{dihedral}}$;非局部项$E_{\mathrm{vdW}}+E_{\mathrm{electro}}$建模非共价键的长程相互作用,包括依赖于原子间距离的范德华力与静电作用。受此启发,我们在PAMNet中也解耦了这两类相互作用的建模:对于局部相互作用,可根据任务需求通过化学键或较小截断距离内的邻近节点定义;对于包含局部与非局部作用的全局相互作用,则通过较大截断距离内的邻近节点定义。每种相互作用类型均使用一个图层表示,其中原子作为节点、相互作用作为边。最终由共享原子群的两层图$G={G_{global},G_{local}}$构成的多重图,即可表征原始3D分子结构 (图 1a)。
Message passing modules. To update the node embeddings in the multiplex graph $G$ , we design two message passing modules that incorporate geometric information: Global Message Passing and Local Message Passing for updating the node embeddings in $G_{g l o b a l}$ and $G_{l o c a l}$ , respectively (Fig. 1b). These message passing modules are inspired by physical principles from molecular mechanics (Fig. 1c): When modeling the molecular energy $E$ , the terms for local interactions require geometric information including inter atomic distances (bond lengths) and angles (bond angles and dihedral angles), while the terms for non-local interactions only require inter atomic distances as geometric information. The message passing modules in PAMNet also use geometric information in this way when modeling these interactions (Fig. 1b and Fig. 1e). Specifically, we capture the pairwise distances and angles contained within up to two-hop neighborhoods (Fig. 1d). The Local Message Passing requires the related adjacency matrix $\mathbf{A}{l o c a l}$ , pairwise distances $d_{l o c a l}$ and angles $\theta_{l o c a l}$ , while the Global Message Passing only needs the related adjacency matrix $\mathbf{A}{g l o b a l}$ and pairwise distances $d_{g l o b a l}$ . Each message passing module then learns the node embeddings $\mathbf{z}{g}$ or $\mathbf{z}{l}$ in $G_{g l o b a l}$ and $G_{l o c a l}$ , respectively.
消息传递模块。为了更新多重图 $G$ 中的节点嵌入,我们设计了两个结合几何信息的消息传递模块:全局消息传递和局部消息传递,分别用于更新 $G_{global}$ 和 $G_{local}$ 中的节点嵌入 (图 1b)。这些消息传递模块的灵感来自分子力学的物理原理 (图 1c):在建模分子能量 $E$ 时,局部相互作用项需要包括原子间距离 (键长) 和角度 (键角和二面角) 在内的几何信息,而非局部相互作用项仅需原子间距离作为几何信息。PAMNet 中的消息传递模块在建模这些相互作用时也以类似方式使用几何信息 (图 1b 和图 1e)。具体而言,我们捕获了最多两跳邻域内的成对距离和角度 (图 1d)。局部消息传递需要相关的邻接矩阵 $\mathbf{A}{local}$、成对距离 $d_{local}$ 和角度 $\theta_{local}$,而全局消息传递仅需相关的邻接矩阵 $\mathbf{A}{global}$ 和成对距离 $d_{global}$。每个消息传递模块随后分别学习 $G_{global}$ 和 $G_{local}$ 中的节点嵌入 $\mathbf{z}{g}$ 或 $\mathbf{z}_{l}$。
For the operations in our message passing modules, they can preserve different symmetries: E(3)-invariance and E(3)- e qui variance, which contain essential inductive bias incorporated by GNNs when dealing with graphs with geometric information8. E(3)-invariance is preserved when predicting E(3)-invariant scalar quantities like energies, which remain unchanged when the original molecular structure undergoes any E(3) transformation including rotation, translation, and reflection. To preserve E(3)-invariance, the input node embeddings $\pmb{h}$ and geometric features are all E(3)-invariant. To update these features, PAMNet utilizes operations that can preserve the invariance. In contrast, E(3)-e qui variance is preserved when predicting E(3)-e qui variant vectorial quantities like dipole moment, which will change according to the same transformation applied to the original molecular structure through E(3) transformation. To preserve E(3)-e qui variance, an extra associated geometric vector $\vec{\nu}\in\mathbb{R}^{3}$ is defined for each node. These geometric vectors are updated by operations inspired by quantum mechanics 22, allowing for the learning of E(3)-e qui variant vectorial representations. More details about the explanations of E(3)-invariance, E(3)-e qui variance, and our operations can be found in Methods.
在我们的消息传递模块中,操作可以保持不同的对称性:E(3)不变性 (E(3)-invariance) 和 E(3)等变性 (E(3)-equivariance),这些对称性包含了图神经网络 (GNN) 在处理具有几何信息的图时引入的关键归纳偏置 [8]。当预测能量等 E(3)不变标量时,E(3)不变性得以保持,这些标量在原始分子结构经历任何 E(3)变换(包括旋转、平移和反射)时保持不变。为了保持 E(3)不变性,输入的节点嵌入 $\pmb{h}$ 和几何特征都是 E(3)不变的。PAMNet 使用能够保持这种不变性的操作来更新这些特征。相比之下,当预测偶极矩等 E(3)等变矢量时,E(3)等变性得以保持,这些矢量会根据通过 E(3)变换应用于原始分子结构的相同变换而改变。为了保持 E(3)等变性,每个节点额外定义了一个关联的几何向量 $\vec{\nu}\in\mathbb{R}^{3}$。这些几何向量通过受量子力学启发的操作 [22] 进行更新,从而学习 E(3)等变的矢量表示。关于 E(3)不变性、E(3)等变性及我们操作的更多细节,请参阅方法部分。
Fusion module. After updating the node embeddings $\mathbf{z}{g}$ or $\mathbf{z}{l}$ of the two layers in the multiplex graph $G$ , we design a fusion module with a two-step pooling process to combine $\mathbf{z}{g}$ and $\mathbf{z}{l}$ for downstream tasks (Figure 1b). In the first step, we design an attention pooling module based on attention mechanism 23 for each hidden layer $t$ in PAMNet. Since $G_{g l o b a l}$ and $G_{l o c a l}$ contains the same set of nodes ${N}$ , we apply the attention mechanism to each node $n\in{N}$ to learn the attention weights $(\alpha_{g}^{t}$ and $\alpha_{l}^{t}$ ) between the node embeddings of $n$ in $G_{g l o b a l}$ and $G_{l o c a l}$ , which are $\mathbf{z}{g}^{t}$ and $\mathbf{z}{l}^{t}$ . Then the attention weights are treated as the importance of $\pmb{z}{g}^{t}$ and $\mathbf{z}_{l}^{t}$ to compute the combined node embedding $\mathbf{z}^{t}$ in each hidden layer $t$ based on a weighted summation (Figure 1e). In the second step, the ${\bf z}^{t}$ of all hidden layers are summed together to compute the node embeddings of the original input. If a graph embedding is desired, we compute it using an average or a summation of the node embeddings.
融合模块。在更新多重图 $G$ 中两个层的节点嵌入 $\mathbf{z}{g}$ 或 $\mathbf{z}{l}$ 后,我们设计了一个包含两步池化过程的融合模块,将 $\mathbf{z}{g}$ 和 $\mathbf{z}{l}$ 结合用于下游任务 (图 1b)。第一步,我们为 PAMNet 中的每个隐藏层 $t$ 设计了一个基于注意力机制 [23] 的注意力池化模块。由于 $G_{global}$ 和 $G_{local}$ 包含相同的节点集 ${N}$,我们对每个节点 $n\in{N}$ 应用注意力机制,学习 $n$ 在 $G_{global}$ 和 $G_{local}$ 中的节点嵌入 $\mathbf{z}{g}^{t}$ 和 $\mathbf{z}{l}^{t}$ 之间的注意力权重 $(\alpha_{g}^{t}$ 和 $\alpha_{l}^{t})$。然后将注意力权重作为 $\pmb{z}{g}^{t}$ 和 $\mathbf{z}_{l}^{t}$ 的重要性,基于加权求和计算每个隐藏层 $t$ 中的组合节点嵌入 $\mathbf{z}^{t}$ (图 1e)。第二步,将所有隐藏层的 ${\bf z}^{t}$ 相加,计算原始输入的节点嵌入。如果需要图嵌入,我们使用节点嵌入的平均值或求和来计算。
Results and discussion
结果与讨论
In this section, we will demonstrate the performance of our proposed PAMNet regarding two aspects: accuracy and efficiency. Accuracy denotes how well the model performs measured by the metrics corresponding to a given task. Efficiency denotes the memory consumed and the inference time spent by the model.
在本节中,我们将从准确性和效率两个方面展示所提出的PAMNet性能。准确性指模型在给定任务对应指标下的表现优劣,效率则涉及模型消耗的内存及推理耗时。
Performance of PAMNet regarding accuracy
PAMNet在准确率方面的表现
Small molecule property prediction. To evaluate the accuracy of PAMNet in learning representations of small 3D molecules, we choose QM9, which is a widely used benchmark for the prediction of 12 molecular properties of around 130k small organic molecules with up to 9 non-hydrogen atoms24. Mean absolute error (MAE) and mean standardized MAE (std. MAE)15 are used for quantitative evaluation of the target properties. Besides evaluating the original PAMNet which captures geometric information within two-hop neighborhoods of each node, we also develop a "simple" PAMNet, called PAMNet-s, that utilizes only the geometric information within one-hop neighborhoods. The PAMNet models are compared with several state-of-the-art models including SchNet13, PhysNet14, $\mathbf{M}\mathbf{G}\mathbf{CN}^{25}$ , $\mathrm{PaiNN}^{26}$ , DimeNet $++^{16}$ , and Sphere Net 27. More details of the experiments can be found in Methods and Supplementary Information.
小分子性质预测。为了评估PAMNet在学习3D小分子表征方面的准确性,我们选择了QM9数据集,该数据集广泛用于预测约13万个最多含9个非氢原子的小有机分子的12种分子性质[24]。我们采用平均绝对误差(MAE)和标准化平均绝对误差(std. MAE)[15]对目标性质进行定量评估。除了评估原始PAMNet(捕获每个节点两跳邻域内的几何信息)外,我们还开发了仅利用一跳邻域几何信息的简化版PAMNet(称为PAMNet-s)。PAMNet模型与多个前沿模型进行了对比,包括SchNet[13]、PhysNet[14]、$\mathbf{M}\mathbf{G}\mathbf{CN}^{25}$、$\mathrm{PaiNN}^{26}$、DimeNet$++^{16}$以及SphereNet[27]。实验详情请参阅方法与补充信息部分。
We compare the performance of PAMNet with those of the baseline models mentioned above on QM9, as shown in Table 1. PAMNet achieves 4 best and 6 second-best results among all 12 properties, while PAMNet-s achieves 3 second-best results. When evaluating the overall performance using the std. MAE across all properties, PAMNet and PAMNet-s rank 1 and 2 among all models with $10%$ and $5%$ better std. MAE than the third-best model (SphereNet), respectively. From the results, we can observe that the models incorporating only atomic pairwise distance $d$ as geometric information like SchNet, PhysNet, and MGCN generally perform worse than those models incorporating more geometric information like PaiNN, DimeNet++, SphereNet, and our PAMNet. Besides, PAMNet-s which captures geometric information only within one-hop neighborhoods performs worse than PAMNet which considers two-hop neighborhoods. These show the importance of capturing rich geometric information when representing 3D small molecules. The superior performance of PAMNet models demonstrates the power of our separate modeling of different interactions in molecules and the effectiveness of the message passing modules designed.
我们在QM9数据集上将PAMNet与上述基线模型的性能进行比较,结果如表1所示。PAMNet在全部12个性质中取得4项最优和6项次优结果,而PAMNet-s获得3项次优成绩。当采用所有性质的标准MAE(std. MAE)评估整体性能时,PAMNet和PAMNet-s分别以比第三名模型(SphereNet)优$10%$和$5%$的标准MAE位列第一和第二。结果表明,仅采用原子对距离$d$作为几何信息的模型(如SchNet、PhysNet和MGCN)普遍逊于整合更多几何信息的模型(如PaiNN、DimeNet++、SphereNet及我们的PAMNet)。此外,仅捕捉单跳邻域几何信息的PAMNet-s性能低于考虑双跳邻域的PAMNet,这印证了在表示3D小分子时捕获丰富几何信息的重要性。PAMNet模型的卓越性能验证了我们分离建模分子内不同相互作用的有效性,以及所设计消息传递模块的优越性。
Table 1. Performance comparison on QM9. The best results are marked in bold and the second-best results with underline.
表 1: QM9 上的性能对比。最佳结果以粗体标出,次优结果以下划线标出。
| 属性 | 单位 | SchNet | PhysNet | MGCN | PaiNN | DimeNet++ | SphereNet | PAMNet-s | PAMNet |
|---|---|---|---|---|---|---|---|---|---|
| μ | mD | 21 | 52.9 | 56 | 12 | 29.7 | 24.5 | 11.3 | 10.8 |
| a | 0.124 | 0.0615 | 0.030 | 0.045 | 0.0435 | 0.0449 | 0.0466 | 0.0447 | |
| EHOMO | meV | 47 | 32.9 | 42.1 | 27.6 | 24.6 | 22.8 | 23.9 | 22.8 |
| ELUMO | meV | 39 | 24.7 | 57.4 | 20.4 | 19.5 | 18.9 | 20.0 | 19.2 |
| meV | 74 | 42.5 | 64.2 | 45.7 | 32.6 | 31.1 | 32.4 | 31.0 | |
| (R2) | a | 0.158 | 0.765 | 0.11 | 0.066 | 0.331 | 0.268 | 0.094 | 0.093 |
| ZPVE | meV | 1.616 | 1.39 | 1.12 | 1.28 | 1.21 | 1.12 | 1.24 | 1.17 |
| Uo | meV | 12 | 8.15 | 12.9 | 5.85 | 6.32 | 6.26 | 6.05 | 5.90 |
| n | meV | 12 | 8.34 | 14.4 | 5.83 | 6.28 | 6.36 | 6.08 | 5.92 |
| H | meV | 12 | 8.42 | 16.2 | 5.98 | 6.53 | 6.33 | 6.19 | 6.04 |
| G | meV | 13 | 9.40 | 14.6 | 7.35 | 7.56 | 7.78 | 7.34 | 7.14 |
| 43 | cal molK | 0.034 | 0.0280 | 0.038 | 0.024 | 0.0230 | 0.0215 | 0.0234 | 0.0231 |
| std. MAE | % | 1.78 | 1.37 | 1.89 | 1.01 | 0.98 | 0.91 | 0.87 | 0.83 |
When predicting dipole moment $\mu$ as a scalar value, which is originally an E(3)-e qui variant vectorial property $\vec{\mu}$ , PAMNet preserves the E(3)-e qui variance to directly predict $\vec{\mu}$ first and then takes the magnitude of $\vec{\mu}$ as the final prediction. As a result, PAMNet and PAMNet-s all get lower MAE ( $10.8~\mathrm{mD}$ and $11.3\mathrm{mD},$ ) than the previous best result $\mathrm{\Delta}12\mathrm{mD},$ ) achieved by PaiNN, which is a GNN with e qui variant operations for predicting vectorial properties. Note that the remaining baselines all directly predict dipole moment as a scalar property by preserving invariance. We also examine that by preserving invariance in PAMNet and directly predicting dipole moment as a scalar property, the MAE $(24.0\mathrm{mD})$ is much higher than the e qui variant version. These results demonstrate that preserving e qui variance is more helpful than preserving invariance for predicting dipole moments.
在预测偶极矩$\mu$这一标量值时(其本质是E(3)-等变的矢量属性$\vec{\mu}$),PAMNet通过保持E(3)-等变性直接预测$\vec{\mu}$,再取其模长作为最终预测值。因此PAMNet与PAMNet-s的MAE($10.8~\mathrm{mD}$和$11.3\mathrm{mD}$)均低于此前由PaiNN(一种用于预测矢量属性的等变图神经网络)取得的最佳结果($\mathrm{\Delta}12\mathrm{mD}$)。需注意其他基线方法均通过保持不变性直接将偶极矩作为标量属性预测。实验表明,若在PAMNet中保持不变性并直接预测标量偶极矩,其MAE($24.0\mathrm{mD}$)将显著高于等变版本。这些结果证明对于偶极矩预测,保持等变性比保持不变性更具优势。
RNA 3D structure prediction. Besides small molecules, we further apply PAMNet to predict RNA 3D structures for evaluating the accuracy of PAMNet in learning representations of 3D macromolecules. Following the previous works28–30, we refer the prediction to be the task of identifying accurate structural models of RNA from less accurate ones: Given a group of candidate 3D structural models generated based on an RNA sequence, a desired model that serves as a scoring function needs to distinguish accurate structural models among all candidates. We use the same datasets as those used $\mathrm{in}^{30}$ , which include a dataset for training and a benchmark for evaluation. The training dataset contains 18 relatively older and smaller RNA molecules experimentally determined 31. Each RNA is used to generate 1000 structural models via the Rosetta FARFAR2 sampling method29. The benchmark for evaluation contains relatively newer and larger RNAs, which are the first 21 RNAs in the RNA-Puzzles structure prediction challenge 32. Each RNA is used to generate at least 1500 structural models using FARFAR2, where $1%$ of the models are near-native (i.e., within a RMSD of the experimentally determined native structure). In practice, each scoring function predicts the root mean square deviation (RMSD) from the unknown true structure for each structural model. A lower RMSD would suggest a more accurate structural model predicted. We compare PAMNet with four state-of-the-art baselines: ARES30, Rosetta (2020 version)29, $\mathrm{RASP}^{33}$ , and 3 d RNA score 28. Among the baselines, only ARES is a deep learning-based method, and is a GNN using e qui variant operations. More details of the experiments are introduced in Methods and Supplementary Information.
RNA 3D结构预测。除小分子外,我们进一步将PAMNet应用于RNA三维结构预测,以评估其学习大分子三维表征的准确性。沿袭前人研究[28-30],我们将该预测任务定义为从低精度模型中识别准确RNA结构模型:给定基于RNA序列生成的一组候选三维结构模型,需要作为评分函数的理想模型从所有候选中区分出准确结构模型。我们采用与文献[30]相同的数据集,包含训练集和评估基准集。训练集包含18个通过实验确定的较早期小型RNA分子[31],每个RNA通过Rosetta FARFAR2采样方法[29]生成1000个结构模型。评估基准集包含较新的大型RNA分子,取自RNA-Puzzles结构预测挑战赛[32]前21个RNA,每个RNA使用FARFAR2生成至少1500个结构模型,其中1%为近天然构象(即与实验测定天然结构的RMSD差值在2Å以内)。实际操作中,每个评分函数预测各结构模型与未知真实结构的均方根偏差(RMSD),RMSD值越低表明预测的结构模型越精确。我们将PAMNet与四种前沿基线方法对比:ARES[30]、Rosetta(2020版)[29]、RASP[33]和3dRNAscore[28]。其中仅ARES是基于深度学习的方法,且为使用等变操作的图神经网络(GNN)。实验细节详见方法与补充信息部分。
On the RNA-Puzzles benchmark for evaluation, PAMNet significantly outperforms all other four scoring functions as shown in Figure 2. When comparing the best-scoring structural model of each RNA (Figure 2a), the probability of the model to be near-native $\mathrm{\Delta}<2\mathrm{\mathring{A}}$ RMSD from the native structure) is $90%$ when using PAMNet, compared with 62, 43, 33, and $5%$ for ARES, Rosetta, RASP, and 3dRNAscore, respectively. As for the 10 best-scoring structural models of each RNA (Figure 2b), the probability of the models to include at least one near-native model is $90%$ when using PAMNet, compared with 81, 48, 48, and $33%$ for ARES, Rosetta, RASP, and 3dRNAscore, respectively. When comparing the rank of the best-scoring near-native structural model of each RNA (Figure 2c), the geometric mean of the ranks across all RNAs is 1.7 for PAMNet, compared with 3.6, 73.0, 26.4, and 127.7 for ARES, Rosetta, RASP, and 3dRNAscore, respectively. The lower mean rank of PAMNet indicates that less effort is needed to go down the ranked list of PAMNet to include one near-native structural model. A more detailed analysis of the near-native ranking task can be found in Supplementary Figure S1.
在RNA-Puzzles评估基准上,PAMNet显著优于其他四种评分函数,如图2所示。当比较每个RNA的最佳评分结构模型时(图2a),使用PAMNet时模型接近天然结构(RMSD $\mathrm{\Delta}<2\mathrm{\mathring{A}}$)的概率为$90%$,而ARES、Rosetta、RASP和3dRNAscore分别为62%、43%、33%和$5%$。对于每个RNA的10个最佳评分结构模型(图2b),使用PAMNet时包含至少一个接近天然模型的概率为$90%$,而ARES、Rosetta、RASP和3dRNAscore分别为81%、48%、48%和$33%$。在比较每个RNA最佳评分的接近天然结构模型的排名时(图2c),PAMNet在所有RNA中的排名几何平均值为1.7,而ARES、Rosetta、RASP和3dRNAscore分别为3.6、73.0、26.4和127.7。PAMNet较低的排名均值表明,在PAMNet的排名列表中需要更少的努力即可包含一个接近天然的结构模型。关于接近天然排名任务的更详细分析可参见补充图S1。
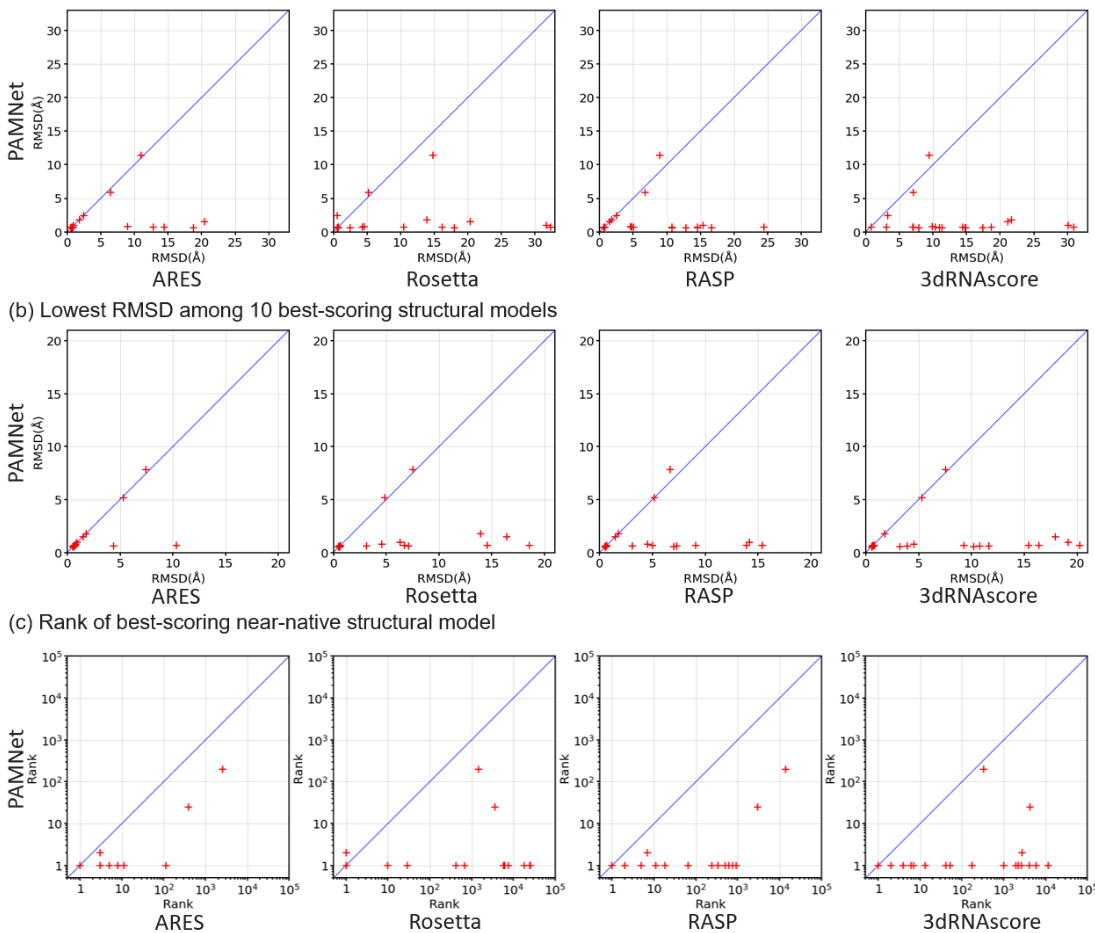
Figure 2. Performance comparison on RNA-Puzzles. Given a group of candidate structural models for each RNA, we rank the models using PAMNet and the other four leading scoring functions for comparison. Each cross in the figures corresponds to one RNA. (a) The best-scoring structural model of each RNA predicted by the scoring functions is compared. PAMNet in general identifies more accurate models (with lower RMSDs from the native structure) than those decided by the other scoring functions. (b) Comparison of the 10 best-scoring structural models. The identification s of PAMNet contain accurate models more frequently than those from other scoring functions. (c) The rank of the best-scoring near-native structural model for each RNA is used for comparison. PAMNet usually performs better than the other scoring functions by having a lower rank.
图 2: RNA-Puzzles性能对比。针对每组RNA候选结构模型,我们使用PAMNet和其他四种领先的评分函数进行模型排序。图中每个十字标记对应一个RNA。(a) 对比各评分函数预测的每个RNA最佳结构模型。PAMNet通常能识别出比其他评分函数更精确(与天然结构RMSD值更低)的模型。(b) 前10个最佳评分结构模型对比。PAMNet的识别结果比其他评分函数更频繁包含精确模型。(c) 对比每个RNA最佳近天然结构模型的排名。PAMNet通常通过获得更低排名而优于其他评分函数。
Protein-ligand binding affinity prediction. In this experiment, we evaluate the accuracy of PAMNet in representing the complexes that contain both small molecules and macromolecules. We use PDBbind, which is a well-known public database of experimentally measured binding affinities for protein-ligand complexes 34. The goal is to predict the binding affinity of each complex based on its 3D structure. We use the PDBbind v2016 dataset and preprocess each original complex to a structure that contains around 300 non hydrogen atoms on average with only the ligand and the protein residues within around it. To comprehensively evaluate the performance, we use Root Mean Square Error (RMSE), Mean Absolute Error (MAE), Pearson’s correlation coefficient (R) and the standard deviation (SD) in regression following 18. PAMNet is compared with various comparative methods including machine learning-based methods (LR, SVR, and RF-Score35), CNN-based methods (Pafnucy36 and $\mathrm{{OnionNet}}^{37}$ ), and GNN-based methods (GraphDTA38, $\mathrm{SGCN}^{39}$ , GNN-DTI40, D-MPNN12, $\mathrm{MAT^{41}}$ , DimeNet15, $\mathrm{{CMPNN^{42}}}$ , and $\mathrm{SIGN^{18}}$ ). More details of the experiments are provided in Methods and Supplementary Information.
蛋白质-配体结合亲和力预测。在本实验中,我们评估了PAMNet在表征包含小分子和大分子的复合物时的准确性。我们使用PDBbind,这是一个著名的公共数据库,包含实验测量的蛋白质-配体复合物结合亲和力[34]。目标是根据每个复合物的3D结构预测其结合亲和力。我们使用PDBbind v2016数据集,并将每个原始复合物预处理为平均包含约300个非氢原子的结构,仅保留配体及其周围范围内的蛋白质残基。为了全面评估性能,我们采用均方根误差(RMSE)、平均绝对误差(MAE)、皮尔逊相关系数(R)和回归中的标准差(SD)作为指标[18]。PAMNet与多种对比方法进行比较,包括基于机器学习的方法(LR、SVR和RF-Score[35])、基于CNN的方法(Pafnucy[36]和$\mathrm{{OnionNet}}^{37}$)以及基于GNN的方法(GraphDTA[38]、$\mathrm{SGCN}^{39}$、GNN-DTI[40]、D-MPNN[12]、$\mathrm{MAT^{41}}$、DimeNet[15]、$\mathrm{{CMPNN^{42}}}$和$\mathrm{SIGN^{18}}$)。实验的更多细节见方法和补充信息。
We list the results of all models and compare their performance in Table 2 and Supplementary Table S1. PAMNet achieves the best performance regarding all 4 evaluation metrics in our experiment. When compared with the second-best model, SIGN, our PAMNet performs significantly better with p-value $<0.05$ . These results clearly demonstrate the accuracy of our model when learning representations of 3D macromolecule complexes.
我们在表2和补充表S1中列出了所有模型的结果并比较了它们的性能。PAMNet在我们的实验中实现了所有4个评估指标的最佳性能。与排名第二的模型SIGN相比,我们的PAMNet表现显著更优(p值$<0.05$)。这些结果清楚地证明了我们的模型在学习3D大分子复合物表征时的准确性。
In general, we find that the models with explicitly encoded 3D geometric information like DimeNet, SIGN, and our PAMNet outperform the other models without the information directly encoded. An exception is that DimeNet cannot beat CMPNN. This might be because DimeNet is domain-specific and is originally designed for small molecules rather than macromolecule
总体而言,我们发现显式编码3D几何信息的模型(如DimeNet、SIGN和我们的PAMNet)性能优于未直接编码该信息的其他模型。例外情况是DimeNet未能超越CMPNN,这可能是因为DimeNet具有领域特异性,其最初设计针对小分子而非大分子。
Table 2. Performance comparison on PDBbind. We report the averaged results together with standard deviations. For the evaluation metrics, $\downarrow$ denotes the lower the better, while $\uparrow$ denotes the higher the better. The best results are marked in bold and the second-best results with underline.
表 2: PDBbind 上的性能对比。我们报告了平均值和标准差。对于评估指标,$\downarrow$ 表示越低越好,而 $\uparrow$ 表示越高越好。最佳结果以粗体标出,次佳结果以下划线标出。
| 模型类型 | 模型 | RMSE↓ | MAE← | SD← | R↑ |
|---|---|---|---|---|---|
| 基于机器学习 | LR | 1.675 (0.000) | 1.358 (0.000) | 1.612 (0.000) | 0.671 (0.000) |
| SVR | 1.555 (0.000) | 1.264 (0.000) | 1.493 (0.000) | 0.727 (0.000) | |
| RF-Score | 1.446 (0.008) | 1.161 (0.007) | 1.335 (0.010) | 0.789 (0.003) | |
| 基于 CNN | Pafnucy | 1.585 (0.013) | 1.284 (0.021) | 1.563 (0.022) | 0.695 (0.011) |
| OnionNet | 1.407 (0.034) | 1.078 (0.028) | 1.391 (0.038) | 0.768 (0.014) | |
| 基于 GNN | GraphDTA | 1.562 (0.022) | 1.191 (0.016) | 1.558 (0.018) | 0.697 (0.008) |
| SGCN | 1.583 (0.033) | 1.250 (0.036) | 1.582 (0.320) | 0.686 (0.015) | |
| GNN-DTI | 1.492 (0.025) | 1.192 (0.032) | 1.471 (0.051) | 0.736 (0.021) | |
| D-MPNN | 1.493 (0.016) | 1.188 (0.009) | 1.489 (0.014) | 0.729 (0.006) | |
| MAT | 1.457 (0.037) | 1.154 (0.037) | 1.445 (0.033) | 0.747 (0.013) | |
| DimeNet | 1.453 (0.027) | 1.138 (0.026) | 1.434 (0.023) | 0.752 (0.010) | |
| CMPNN | 1.408 (0.028) | 1.117 (0.031) | 1.399 (0.025) | 0.765 (0.009) | |
| SIGN | 1.316 (0.031) | 1.027 (0.025) | 1.312 (0.035) | 0.797 (0.012) | |
| 我们的方法 | PAMNet | 1.263 (0.017) | 0.987 (0.013) | 1.261 (0.015) | 0.815 (0.005) |
| 数据集 | 模型 | 内存 (GB) | 推理时间 (s) |
|---|---|---|---|
| QM9 | DimeNet++ | 21.1 | 11.3 |
| QM9 | SphereNet | 22.7 | 11.1 |
| QM9 | PAMNet-s | 6.0 | 7.3 |
| QM9 | PAMNet | 6.2 | 11.0 |
| QM9 | ARES | 13.5 | 2.1 |
| RNA-Puzzles | PAMNet | 7.8 | 0.6 |
| RNA-Puzzles | |||
| PDBbind | SIGN | 19.7 | 12.0 |
| PDBbind | PAMNet | 13.1 | 1.8 |
Table 3. Results of efficiency evaluation. We compare PAMNet with the best-performed baselines in each of the three tasks regarding memory consumption and inference time. The most efficient results are marked in bold.
表 3: 效率评估结果。我们将 PAMNet 与三项任务中每项任务表现最佳的基线在内存消耗和推理时间方面进行比较。最高效的结果以粗体标出。
complexes. In contrast, our proposed PAMNet is more flexible to learn representations for various types of molecular systems. The superior performance of PAMNet for predicting binding affinity relies on the separate modeling of local and non-local interactions. For protein-ligand complexes, the local interactions mainly capture the interactions inside the protein and the ligand, while the non-local interactions can capture the interactions between protein and ligand. Thus PAMNet is able to effectively handle diverse interactions and achieve accurate results.
相比之下,我们提出的PAMNet能更灵活地学习各类分子系统的表征。PAMNet在预测结合亲和力方面的卓越性能依赖于其对局部与非局部相互作用的分别建模。对于蛋白质-配体复合物,局部相互作用主要捕获蛋白质和配体内部的相互作用,而非局部相互作用则能捕获蛋白质与配体间的相互作用。因此,PAMNet能有效处理多样化的相互作用并获得精确结果。
Performance of PAMNet regarding efficiency
PAMNet 的效率表现
To evaluate the efficiency of PAMNet, we compare it to the best-performed baselines in each task regarding memory consumption and inference time and summarize the results in Table 3. Theoretically, DimeNet++, SphereNet, and SIGN all require $O(N k^{2})$ messages in message passing, while our PAMNet requires ${\cal O}(N(k_{g}+{k_{l}}^{2}))$ ) messages instead, where $N$ is the number of nodes, $k$ is the average degree in a graph, $k_{g}$ and $k_{l}$ denotes the average degree in $G_{g}$ and $G_{l}$ in the corresponding multiplex graph $G$ . When $k_{g}\sim k$ and $k_{l}\ll k_{g}$ , PAMNet is much more efficient regarding the number of messages involved. A more detailed analysis of computational complexity is included in Methods. Based on the results in Table 3 empirically, we find PAMNet models all require less memory consumption and inference time than the best-performed baselines in all three tasks, which matches our theoretical analysis. We also compare the memory consumption when using a different largest cutoff distance $d$ of the related models in Figure 3. From the results, we observe that the memory consumed by DimeNet and SIGN increases much faster than PAMNet when $d$ increases. When fixing as an example, PAMNet requires $80%$ and $71%$ less memory than DimeNet and SIGN, respectively. Thus PAMNet is much more memory-efficient and is able to capture longer-range interactions than these baselines with restricted resources. The efficiency of PAMNet models comes from the separate modeling of local and non-local interactions in 3D molecular structures. By doing so, when modeling the non-local interactions, which make up the majority of all interactions, we utilize a relatively efficient message passing scheme that only encodes pairwise distances $d$ as the geometric information. Thus when compared with the models that require more comprehensive geometric information when modeling all interactions, PAMNet significantly reduces the computationally expensive operations. More information about the details of experimental settings is included in Methods.
为了评估PAMNet的效率,我们将其与各任务中表现最佳的基线方法在内存消耗和推理时间方面进行比较,并将结果总结在表3中。理论上,DimeNet++、SphereNet和SIGN在消息传递中都需要$O(N k^{2})$条消息,而我们的PAMNet仅需${\cal O}(N(k_{g}+{k_{l}}^{2}))$条消息。其中$N$表示节点数量,$k$为图的平均度数,$k_{g}$和$k_{l}$分别代表多重图$G$中$G_{g}$和$G_{l}$的平均度数。当$k_{g}\sim k$且$k_{l}\ll k_{g}$时,PAMNet在消息数量方面效率显著更高。关于计算复杂度的详细分析见方法部分。
根据表3的实验结果,我们发现PAMNet模型在所有三项任务中的内存消耗和推理时间均低于表现最佳的基线方法,这与理论分析一致。图3展示了不同最大截断距离$d$下相关模型的内存消耗对比。结果显示,当$d$增大时,DimeNet和SIGN的内存消耗增速远高于PAMNet。以固定为例,PAMNet的内存需求分别比DimeNet和SIGN减少80%和71%。因此PAMNet不仅内存效率更高,还能在有限资源下捕捉更长程的相互作用。
PAMNet的高效性源于其对3D分子结构中局部与非局部相互作用的分别建模。通过这种方式,在建模占主导地位的非局部相互作用时,我们采用了仅需编码成对距离$d$作为几何信息的高效消息传递方案。相比那些建模所有相互作用时需要更全面几何信息的模型,PAMNet显著减少了计算开销较大的操作。实验设置的详细信息见方法部分。
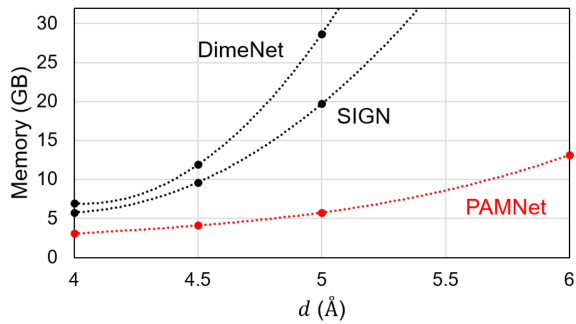
Figure 4. Ablation study of PAMNet. We compare the variants with the original PAMNet and report the differences.
图 4: PAMNet消融实验。我们将变体与原始PAMNet进行比较并报告差异。
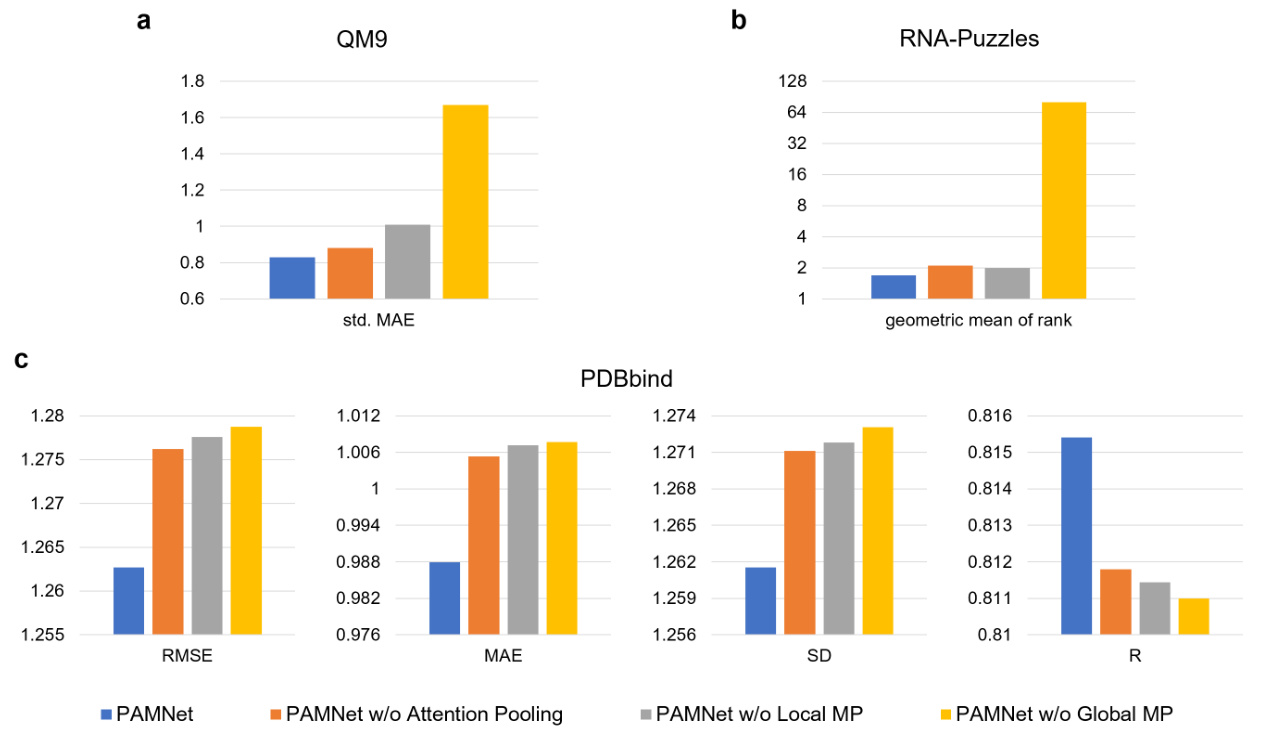
Figure 3. Memory consumption vs. the largest cutoff distance $d$ on PDBbind. We compare PAMNet with the GNN baselines that also explicitly incorporate the 3D molecular geometric information like pairwise distances and angles.
图 3: PDBbind 数据集上内存消耗与最大截断距离 $d$ 的关系。我们将 PAMNet 与同样显式结合 3D 分子几何信息 (如原子对距离和角度) 的 GNN 基线模型进行对比。
All components in PAMNet contribute to the performance
PAMNet中的所有组件都对性能有所贡献
To figure out whether all of the components in PAMNet, including the fusion module and the message passing modules, contribute to the performance of PAMNet, we conduct an ablation study by designing PAMNet variants. Without the attention pooling, we use the averaged results from the message passing modules in each hidden layer to build a variant. We also remove either the Local Message Passing or the Global Message Passing for investigation. The performances of all PAMNet variants are evaluated on the three benchmarks. Specifically, the std. MAE across all properties on QM9, the geometric mean of the ranks across all RNAs on RNA-Puzzles, and the four metrics used in the experiment on PDBbind are computed for comparison. The results in Figure 4 show that all variants decrease the performance of PAMNet in the evaluations, which clearly validates the contributions of all those components. Detailed results of the properties on QM9 can be found in Supplementary Table S2.
为了验证PAMNet中所有组件(包括融合模块和消息传递模块)是否都对性能有贡献,我们通过设计PAMNet变体进行了消融实验。去除注意力池化模块后,我们使用各隐藏层消息传递模块的平均结果构建了一个变体。此外,我们还分别移除了局部消息传递(Local Message Passing)和全局消息传递(Global Message Passing)进行测试。所有PAMNet变体的性能均在三个基准数据集上评估:计算QM9所有属性的标准差MAE、RNA-Puzzles中所有RNA的几何平均排名、以及PDBbind实验中使用的四项指标进行对比。图4结果显示,所有变体都会导致PAMNet性能下降,这明确验证了各组件的贡献。QM9各属性的详细结果可参见补充材料表S2。
Analysis of the contribution of local and global interactions
局部与全局交互作用贡献分析
A salient property of PAMNet is the incorporation of the attention mechanism in the fusion module, which takes the importance of node embeddings in $G_{l o c a l}$ and $G_{g l o b a l}$ of $G$ into consideration in learning combined node embeddings. Recall that for each node $n$ in the set of nodes ${N}$ in $G$ , the attention pooling in the fusion module learns the attention weights $\alpha_{l}$ and $\alpha_{g}$ between $n$ ’s node embedding $\mathbf{z}{l}$ in $G_{l o c a l}$ and $n$ ’s node embedding $\mathbf{z}{g}$ in $G_{g l o b a l}$ . $\alpha_{l}$ and $\alpha_{g}$ serve as the importance of $\mathbf{z}{l}$ and $\mathbf{z}{g}$ when computing the combined node embedding $\mathbf{z}$ . To better understand the contribution of $\mathbf{z}{l}$ and $\mathbf{z}{g}$ , we conduct a detailed analysis of the learned attention weights $\alpha_{l}$ and $\alpha_{g}$ in the three tasks we experimented with. Since the node embeddings are directly related to the involved interactions, such analysis can also reveal the contribution of local and global interactions on the predictions in different tasks. In each task, we take an average of all $\alpha_{l}$ or $\alpha_{g}$ to be the overall importance of the corresponding group of interactions. Then we compare the computed average attention weights $\overline{{\alpha_{l}}}$ and $\overline{{\alpha_{g}}}$ and list the results in Table 4. A higher attention weight in each task indicates a stronger contribution of the corresponding interactions on solving the task.
PAMNet的一个显著特性是在融合模块中引入了注意力机制,该机制在学习组合节点嵌入时考虑了节点在$G$的$G_{local}$和$G_{global}$中的重要性。对于$G$中节点集${N}$中的每个节点$n$,融合模块中的注意力池化会学习$n$在$G_{local}$中的节点嵌入$\mathbf{z}{l}$与$G_{global}$中的节点嵌入$\mathbf{z}{g}$之间的注意力权重$\alpha_{l}$和$\alpha_{g}$。$\alpha_{l}$和$\alpha_{g}$在计算组合节点嵌入$\mathbf{z}$时分别表示$\mathbf{z}{l}$和$\mathbf{z}{g}$的重要性。为了更好地理解$\mathbf{z}{l}$和$\mathbf{z}{g}$的贡献,我们对实验中的三个任务所学习到的注意力权重$\alpha_{l}$和$\alpha_{g}$进行了详细分析。由于节点嵌入与涉及的交互直接相关,这种分析还可以揭示局部和全局交互在不同任务预测中的贡献。在每个任务中,我们将所有$\alpha_{l}$或$\alpha_{g}$的平均值作为相应交互组的整体重要性,然后比较计算得到的平均注意力权重$\overline{{\alpha_{l}}}$和$\overline{{\alpha_{g}}}$,并将结果列在表4中。每个任务中较高的注意力权重表明相应交互在解决该任务时的贡献更强。
Table 4. Comparison of the average attention weights $\overline{{\alpha_{l}}}$ and $\overline{{\alpha_{g}}}$ for local and global interactions in attention pooling. The higher attention weight for each target is marked in bold.
表 4: 注意力池化中局部交互 $\overline{{\alpha_{l}}}$ 和全局交互 $\overline{{\alpha_{g}}}$ 的平均注意力权重对比。每个目标的较高注意力权重以粗体标出。
| 注意力权重 | QM9 | RNA-Puzzles | PDBbind | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μ | α | EHOMO | ELUMO | (R²) | ZPVE | Uo | U | H | G | Cv | ||
| α1 | 0.64 | 0.53 | 0.50 | 0.50 | 0.29 | 0.54 | 0.60 | 0.60 | 0.60 | 0.57 | 0.58 | 0.22 |
| Og | 0.36 | 0.47 | 0.50 | 0.50 | 0.71 | 0.46 | 0.40 | 0.40 | 0.40 | 0.43 | 0.42 | 0.78 |
For the targets being predicted in QM9, we find that all of them have $\overline{{\alpha_{l}}}\geq\overline{{\alpha_{g}}}$ except the electronic spatial extent $\left\langle R^{2}\right\rangle$ , indicating a stronger contribution of the local interactions, which are defined by chemical bonds in this task. This may be because QM9 contains small molecules with only up to 9 non-hydrogen atoms, local interactions can capture a considerable portion of all atomic interactions. However, when predicting electronic spatial extent $\left\langle R^{2}\right\rangle$ , we notice that $\overline{{\alpha_{l}}}<\overline{{\alpha_{g}}}$ , which suggests that $\left\langle R^{2}\right\rangle$ is mainly affected by the global interactions that are the pairwise interactions within in this case. This is not surprising since $\left\langle R^{2}\right\rangle$ is the electric field area affected by the ions in the molecule, and is directly related to the diameter or radius of the molecule. Besides, previous study43 has demonstrated that graph properties like diameter and radius cannot be computed by message passing-based GNNs that rely entirely on local information, and additional global information is needed. Thus it is expected that global interactions have a stronger contribution than local interactions on predicting electronic spatial extent.
在QM9中被预测的目标中,我们发现除电子空间范围$\left\langle R^{2}\right\rangle$外,所有目标都满足$\overline{{\alpha_{l}}}\geq\overline{{\alpha_{g}}}$,表明局部相互作用(本任务中由化学键定义)贡献更强。这可能是因为QM9仅包含不超过9个非氢原子的小分子,局部相互作用能捕捉到相当比例的原子间作用。然而在预测电子空间范围$\left\langle R^{2}\right\rangle$时,我们注意到$\overline{{\alpha_{l}}}<\overline{{\alpha_{g}}}$,说明$\left\langle R^{2}\right\rangle$主要受全局相互作用(本案例中定义为内的成对相互作用)影响。这并不意外,因为$\left\langle R^{2}\right\rangle$表示分子中离子影响的电场区域,与分子直径或半径直接相关。此外,先前研究[43]表明,直径和半径等图属性无法通过完全依赖局部信息的基于消息传递的GNN计算,需要额外的全局信息。因此可以预期,全局相互作用对预测电子空间范围的贡献强于局部相互作用。
For the RNA 3D structure prediction on RNA-Puzzles and the protein-ligand binding affinity prediction on PDBbind, we find $\overline{{\alpha_{l}}}<\overline{{\alpha_{g}}}$ in both cases, which indicates that global interactions play a more important role than local interactions. It is because the goals of these two tasks highly rely on global interactions, which are necessary for representing the global structure of RNA when predicting RNA 3D structure, and are crucial for capturing the relationships between protein and ligand when predicting binding affinity.
在RNA-Puzzles上的RNA三维结构预测和PDBbind上的蛋白质-配体结合亲和力预测任务中,我们发现两种情况均满足$\overline{{\alpha_{l}}}<\overline{{\alpha_{g}}}$,这表明全局相互作用比局部相互作用起着更关键的作用。这是因为这两个任务的目标高度依赖于全局相互作用:预测RNA三维结构时需要全局相互作用来表征RNA的整体结构,而预测结合亲和力时全局相互作用对捕捉蛋白质与配体间的关系至关重要。
Conclusion
结论
In this work, we tackle the limitations of previous GNNs regarding their limited applicability and inefficiency for representation learning of molecular systems with 3D structures and propose a universal framework, PAMNet, to accurately and efficiently learn the representations of 3D molecules in any molecular system. PAMNet explicitly models local and non-local interaction as well as their combined effects inspired by molecular mechanics. The resulting framework incorporates rich geometric information like distances and angles when modeling local interactions, and avoids using expensive operations on modeling nonlocal interactions. Besides, PAMNet learns the contribution of different interactions to combine the updated node embeddings for the final output. When designing the aforementioned operations in PAMNet, we preserve E(3)-invariance for scalar output and preserve E(3)-e qui variance for vectorial output to enable more applicable cases. In our experiments, we evaluate the performance of PAMNet with state-of-the-art baselines on various tasks involving different molecular systems, including small molecules, RNAs, and protein-ligand complexes. In each task, PAMNet outperforms the corresponding baselines in terms of both accuracy and efficiency. These results clearly demonstrate the generalization power of PAMNet even though non-local interactions in molecules are modeled with only pairwise distances as geometric information.
在本研究中,我们针对现有图神经网络(GNN)在三维结构分子系统表征学习中的适用性局限性和低效问题,提出了一种通用框架PAMNet,能够准确高效地学习任意分子系统中三维分子的表征。PAMNet受分子力学启发,显式建模了局部与非局部相互作用及其协同效应。该框架在建模局部相互作用时整合了距离、角度等丰富几何信息,同时避免在非局部相互作用建模中使用高成本运算。此外,PAMNet通过学习不同相互作用的贡献度来融合更新的节点嵌入以生成最终输出。在设计上述运算时,我们保持了标量输出的E(3)不变性和矢量输出的E(3)等变性,以支持更广泛的应用场景。实验部分,我们在包含小分子、RNA和蛋白质-配体复合物等不同分子系统的多项任务中,将PAMNet与最先进的基线方法进行性能对比。结果表明,PAMNet在所有任务中的准确性和效率均优于对应基线方法。这些发现充分证明了PAMNet的泛化能力——即使仅使用成对距离作为几何信息来建模分子中的非局部相互作用。
An under-investigated aspect of our proposed PAMNet is that PAMNet preserves E(3)-invariance in operations when predicting scalar properties while requiring additional representations and operations to preserve E(3)-e qui variance for vectorial properties. Considering that various e qui variant GNNs have been proposed for predicting either scalar or vectorial properties solely by preserving e qui variance, it would be worth extending the idea in PAMNet to e qui variant GNNs with a potential to further improve both accuracy and efficiency. Another interesting direction is that although we only experiment PAMNet on single-task learning, PAMNet is promising to be used in multi-task learning across diverse tasks that involve molecules of varying sizes and types to gain better generalization. Besides using PAMNet for predicting ph y sio chemical properties of molecules, PAMNet can be used as a universal building block for the representation learning of molecular systems in various molecular science problems. Another promising application of PAMNet is self-supervised learning for molecular systems with few labeled data (e.g., RNA structures). For example, we can use the features in one graph layer to learn properties in another graph layer by utilizing the multiplex nature of PAMNet.
我们提出的PAMNet中一个尚未深入研究的方面是:在预测标量属性时,PAMNet保持了操作的E(3)不变性,同时需要额外的表示和操作来保持矢量属性的E(3)等变性。考虑到目前已提出各种等变图神经网络(GNN)通过保持等变性来单独预测标量或矢量属性,将PAMNet的思想扩展到等变GNN中可能进一步提升准确性和效率。另一个有趣的方向是,尽管我们仅在单任务学习上测试了PAMNet,但它有望应用于跨不同尺寸和类型分子的多任务学习,以获得更好的泛化能力。除了使用PAMNet预测分子的物理化学性质外,它还可作为分子系统表示学习的通用构建模块,应用于各种分子科学问题。PAMNet另一个潜在应用场景是数据标签稀缺的分子系统(如RNA结构)的自监督学习。例如,我们可以利用PAMNet的多重特性,通过一个图层的特征来学习另一个图层的属性。
Methods
方法
Details of PAMNet
PAMNet 详情
In this section, we will describe PAMNet in detail, including the involved features, embeddings, and operations.
在本节中,我们将详细描述PAMNet,包括所涉及的特征、嵌入和操作。
Input features. The input features of PAMNet include atomic features and geometric information as shown in Figure 1b. For atomic features, we use only atomic numbers $Z$ for the tasks on QM9 and RNA-Puzzles following 13–16,30, and use 18 chemical features like atomic numbers, hybridization, aromaticity, partial charge, etc., for the task on PDBbind following 18,36. The atomic numbers $Z$ are represented by randomly initialized, trainable embeddings according to13–16. For geometric information, we capture the needed pairwise distances and angles in the multiplex molecular graph $G$ as shown in Figure 1d. The features (d, $\theta$ ) for the distances and angles are computed with the basis functions $\mathrm{in}^{15}$ to reduce correlations. For the prediction of vectorial properties, we use the atomic position $\vec{r}$ to be the initial associated geometric vector $\vec{\nu}$ of each atom.
输入特征。PAMNet的输入特征包括原子特征和几何信息,如图1b所示。对于原子特征,在QM9和RNA-Puzzles任务中我们仅使用原子序数$Z$(遵循13-16,30),而在PDBbind任务中则采用18种化学特征(如原子序数、杂化类型、芳香性、部分电荷等)(遵循18,36)。原子序数$Z$通过随机初始化、可训练的嵌入向量表示(依据13-16)。对于几何信息,我们在多重分子图$G$中捕获所需的成对距离和角度(如图1d所示)。距离和角度的特征(d, $\theta$)通过15中的基函数计算以减少相关性。对于矢量属性的预测,我们使用原子位置$\vec{r}$作为每个原子的初始关联几何向量$\vec{\nu}$。
Message embeddings. In the message passing scheme7, the update of node embeddings $h$ relies on the passing of the related messages $m$ between nodes. In PAMNet, we define the input message embeddings $m$ of message passing schemes with the following way:
消息嵌入 (Message embeddings) 。在消息传递方案7中,节点嵌入 $h$ 的更新依赖于节点间相关消息 $m$ 的传递。在PAMNet中,我们通过以下方式定义消息传递方案的输入消息嵌入 $m$ :
$$
m_{j i}=\mathbf{MLP}{m}\big([h_{j}|h_{i}|e_{j i}]\big),
$$
$$
m_{j i}=\mathbf{MLP}{m}\big([h_{j}|h_{i}|e_{j i}]\big),
$$
where $i,j\in G_{g l o b a l}$ or $G_{l o c a l}$ are connected nodes that can define a message embedding, MLP denotes the multi-layer perceptron, $|$ denotes the concatenation operation. The edge embedding $e_{j i}$ encodes the corresponding pairwise distance $d$ between node $i,j$ .
其中 $i,j\in G_{g l o b a l}$ 或 $G_{l o c a l}$ 为可定义消息嵌入的相连节点,MLP表示多层感知机 (multi-layer perceptron),$|$ 表示拼接操作。边嵌入 $e_{j i}$ 编码了节点 $i,j$ 间对应的成对距离 $d$。
Global message passing. As depicted in Figure 1e, the Global Message Passing in each hidden layer of PAMNet, which consists of a message block and an update block, updates the node embeddings $h$ in $G_{g l o b a l}$ by using the related adjacency matrix $\mathbf{A}{g l o b a l}$ and pairwise distances $d_{g l o b a l}$ . The message block is defined as below to perform the message passing operation:
全局信息传递。如图 1e 所示,PAMNet 每个隐藏层中的全局信息传递 (Global Message Passing) 由消息块和更新块组成,通过相关邻接矩阵 $\mathbf{A}{g l o b a l}$ 和成对距离 $d_{g l o b a l}$ 更新 $G_{g l o b a l}$ 中的节点嵌入 $h$。消息块定义如下以执行信息传递操作:
$$
\begin{array}{r}{\begin{array}{r}{h_{i}^{t}=h_{i}^{t-1}+\sum_{j\in\mathcal{N}(i)}m_{j i}^{t-1}\odot\phi_{d}(e_{j i}),}\end{array}}\end{array}
$$
$$
\begin{array}{r}{\begin{array}{r}{h_{i}^{t}=h_{i}^{t-1}+\sum_{j\in\mathcal{N}(i)}m_{j i}^{t-1}\odot\phi_{d}(e_{j i}),}\end{array}}\end{array}
$$
where $i,j\in G_{g l o b a l}$ , $\phi_{d}$ is a learnable function, $e_{j i}$ is the embedding of pairwise distance $d$ between node $i,j$ , and $\odot$ denotes the element-wise production. After the message block, an update block is used to compute the node embeddings $h$ for the next layer as well as the output $\mathbf{z}$ for this layer. We define the update block using a stack of three residual blocks, where each residual block consists of a two-layer MLP and a skip connection across the MLP. There is also a skip connection between the input of the message block and the output of the first residual block. After the residual blocks, the updated node embeddings $h$ are passed to the next layer. For the output $\mathbf{z}$ of this layer to be combined in the fusion module, we further use a three-layer MLP to get $\mathbf{z}$ with desired dimension size.
其中 $i,j\in G_{global}$,$\phi_{d}$ 是可学习函数,$e_{ji}$ 表示节点 $i,j$ 之间成对距离 $d$ 的嵌入表示,$\odot$ 表示逐元素乘积。在消息传递块之后,更新块用于计算下一层的节点嵌入 $h$ 以及本层输出 $\mathbf{z}$。我们采用包含三个残差块的堆叠结构定义更新块,每个残差块由两层MLP和跨MLP的跳跃连接组成。消息块输入与第一个残差块输出之间也设有跳跃连接。经过残差块处理后,更新后的节点嵌入 $h$ 将传递至下一层。对于需要融合模块整合的本层输出 $\mathbf{z}$,我们额外使用三层MLP来获得指定维度的 $\mathbf{z}$。
Local message passing. For the updates of node embeddings $h$ in $G_{l o c a l}$ , we incorporate both pairwise distances $d_{l o c a l}$ and angles $\theta_{l o c a l}$ as shown in Figure 1e. To capture $\theta_{l o c a l}$ , we consider up to the two-hop neighbors of each node. In Figure 1d, we show an example of the angles we considered: Some angles are between one-hop edges and two-hop edges (e.g. $\angle i j_{1}k_{1}$ ), while the other angles are between one-hop edges (e.g. $\angle j_{1}i j_{2}$ ). Compared to previous $\mathrm{GNNs}^{15-17}$ that incorporate only part of these angles, our PAMNet is able to encode the geometric information more comprehensively. In the Local Message Passing, we also use a message block and an update block following the design of the Global Message Passing as shown in Figure 1e. However, the message block is defined differently as the one in the Global Message Passing to encode additional angular information:
局部消息传递。对于 $G_{local}$ 中节点嵌入 $h$ 的更新,我们同时结合了成对距离 $d_{local}$ 和角度 $\theta_{local}$ 如图 1e 所示。为捕捉 $\theta_{local}$,我们考虑每个节点的两跳邻域范围。图 1d 展示了所考虑角度的示例:部分角度存在于一跳边与二跳边之间(如 $\angle ij_{1}k_{1}$),另一些则存在于一跳边之间(如 $\angle j_{1}ij_{2}$)。相比仅包含部分角度的先前 $\mathrm{GNNs}^{15-17}$,我们的 PAMNet 能更全面地编码几何信息。局部消息传递同样采用消息块和更新块的设计(如图 1e 所示),但消息块的定义与全局消息传递不同,以编码额外的角度信息:
$$
\begin{array}{r l r}{{m_{j i}^{\prime t-1}=m_{j i}^{t-1}+\sum_{\boldsymbol{j}^{\prime}\in\mathcal{N}(i)\setminus{\boldsymbol{j}}}m_{j^{\prime}i}^{t-1}\odot\phi_{d}(e_{j^{\prime}i})\odot\phi_{\theta}(a_{j^{\prime}i,j i})+\sum_{\boldsymbol{k}\in\mathcal{N}(j)\setminus{i}}m_{k j}^{t-1}\odot\phi_{d}(e_{k j})\odot\phi_{\theta}(a_{k j,j i}),}}\ &{}&{h_{i}^{t}=h_{i}^{t-1}+\sum_{\boldsymbol{j}\in\mathcal{N}(i)}m_{j i}^{t-1}\odot\phi_{d}(e_{j i}),}\end{array}
$$
$$
\begin{array}{r l r}{{m_{j i}^{\prime t-1}=m_{j i}^{t-1}+\sum_{\boldsymbol{j}^{\prime}\in\mathcal{N}(i)\setminus{\boldsymbol{j}}}m_{j^{\prime}i}^{t-1}\odot\phi_{d}(e_{j^{\prime}i})\odot\phi_{\theta}(a_{j^{\prime}i,j i})+\sum_{\boldsymbol{k}\in\mathcal{N}(j)\setminus{i}}m_{k j}^{t-1}\odot\phi_{d}(e_{k j})\odot\phi_{\theta}(a_{k j,j i}),}}\ &{}&{h_{i}^{t}=h_{i}^{t-1}+\sum_{\boldsymbol{j}\in\mathcal{N}(i)}m_{j i}^{t-1}\odot\phi_{d}(e_{j i}),}\end{array}
$$
where $i,j,k\in G_{l o c a l}$ , $e_{j i}$ is the embedding of pairwise distance $d$ between node $i,j,a_{k j,j i}$ is the embedding of angle $\theta_{k j,j i}=\angle k j i$ defined by node $i,j,k$ , and $\phi_{d},\phi_{\theta}$ are learnable functions. In Equation (3), we use two summation terms to separately encode the angles in different hops with the associated pairwise distances to update $m_{j i}$ . Then in Equation (4), the updated message embeddings $m_{j i}^{\prime}$ are used to perform message passing. After the message block, we use the same update block as the one used in the Global Message Passing for updating the learned node embeddings.
其中 $i,j,k\in G_{l o c a l}$,$e_{j i}$ 是节点 $i,j$ 之间成对距离 $d$ 的嵌入表示,$a_{k j,j i}$ 是由节点 $i,j,k$ 定义的角度 $\theta_{k j,j i}=\angle k j i$ 的嵌入表示,$\phi_{d},\phi_{\theta}$ 是可学习函数。在公式 (3) 中,我们使用两个求和项分别编码不同跳数的角度及其关联的成对距离来更新 $m_{j i}$。然后在公式 (4) 中,更新后的消息嵌入 $m_{j i}^{\prime}$ 被用于执行消息传递。消息块之后,我们采用与全局消息传递相同的更新块来更新学习到的节点嵌入。
Fusion module. The fusion module consists of two steps of pooling as shown in Figure 1b. In the first step, attention pooing is utilized to learn the combined embedding $\mathbf{z}^{t}$ based on the output node embeddings $\mathbf{z}{g}^{t}$ and $\mathbf{z}{l}^{t}$ in each hidden layer $t$ . The detailed architecture of attention pooling is illustrated in Figure 1e. We first compute the attention weight $\alpha_{p,i}$ on node $i$ that measures the contribution of the results from plex or graph layer $p\in{g,l}$ in multiplex graph $G$ :
融合模块。该模块包含如图1b所示的两步池化操作。第一步采用注意力池化(attention pooling)技术,基于每个隐藏层$t$中图嵌入$\mathbf{z}{g}^{t}$与局部嵌入$\mathbf{z}{l}^{t}$学习组合嵌入$\mathbf{z}^{t}$。注意力池化架构细节如图1e所示:首先计算节点$i$的注意力权重$\alpha_{p,i}$,该权重衡量多重图$G$中plex层或图层$p\in{g,l}$结果的贡献度:
$$
\alpha_{p,i}^{t}=\frac{\exp(\mathrm{LeakyReLU}(W_{p}^{t}\mathbf{z}{p,i}^{t}))}{\sum_{p}\exp(\mathrm{LeakyReLU}(W_{p}^{t}\mathbf{z}_{p,i}^{t}))},
$$
$$
\alpha_{p,i}^{t}=\frac{\exp(\mathrm{LeakyReLU}(W_{p}^{t}\mathbf{z}{p,i}^{t}))}{\sum_{p}\exp(\mathrm{LeakyReLU}(W_{p}^{t}\mathbf{z}_{p,i}^{t}))},
$$
where $W_{p}^{t}\in\mathbb{R}^{1\times F}$ is a learnable weight matrix different for each hidden layer $t$ and graph layer $p$ , and $F$ is the dimension size of $\mathbf{z}{p,i}^{t}$ . With $\boldsymbol{\alpha}{p,i}^{t}$ , we can compute the combined node embedding $\mathbf{z}_{i}^{t}$ of node $i$ using a weighted summation:
其中 $W_{p}^{t}\in\mathbb{R}^{1\times F}$ 是每个隐藏层 $t$ 和图层 $p$ 独有的可学习权重矩阵,$F$ 表示 $\mathbf{z}{p,i}^{t}$ 的维度大小。通过 $\boldsymbol{\alpha}{p,i}^{t}$ ,我们可以使用加权求和计算节点 $i$ 的组合节点嵌入 $\mathbf{z}_{i}^{t}$:
$$
\begin{array}{r}{\mathbf{z}{i}^{t}=\sum_{p}\alpha_{p,i}^{t}(W_{p}^{'t}\mathbf{z}_{p,i}^{t}),}\end{array}
$$
$$
\begin{array}{r}{\mathbf{z}{i}^{t}=\sum_{p}\alpha_{p,i}^{t}(W_{p}^{'t}\mathbf{z}_{p,i}^{t}),}\end{array}
$$
where $W_{p}^{'t}\in\mathbb{R}^{D\times F}$ is a learnable weight matrix different for each hidden layer $t$ and graph layer $p,D$ is the dimension size of $\mathbf{z}{i}^{t}$ , and $F$ i s the dimension size of $\mathbf{z}_{p,i}^{t}$ .
其中 $W_{p}^{'t}\in\mathbb{R}^{D\times F}$ 是可学习的权重矩阵,每个隐藏层 $t$ 和图层 $p$ 都不同,$D$ 是 $\mathbf{z}{i}^{t}$ 的维度大小,$F$ 是 $\mathbf{z}_{p,i}^{t}$ 的维度大小。
In the second step of the fusion module, we sum the combined node embedding $\mathbf{z}$ of all hidden layers to compute the final node embeddings $y$ . If a graph-level embedding $y$ is desired, we compute as follows:
在融合模块的第二步中,我们对所有隐藏层的组合节点嵌入$\mathbf{z}$进行求和,以计算最终的节点嵌入$y$。如果需要图级嵌入$y$,则按如下方式计算:
$$
\begin{array}{r}{y=\sum_{i=1}^{N}\sum_{t=1}^{T}\mathbf{z}_{i}^{t}.}\end{array}
$$
$$
\begin{array}{r}{y=\sum_{i=1}^{N}\sum_{t=1}^{T}\mathbf{z}_{i}^{t}.}\end{array}
$$
Preservation of E(3)-invariance $\pmb{}$ E(3)-e qui variance. For the operations described above, they preserve the E(3)-invariance of the input atomic features and geometric information and can predict E(3)-invariant scalar properties. To predict E(3)- e qui variant vectorial property $\vec{u}$ , we introduce an associated geometric vector $\vec{\nu}{i}$ for each node $i$ and extend PAMNet to preserve the E(3)-e qui variance for learning $\vec{u}$ . In detail, the associated geometric vector $\vec{\nu}_{i}^{t}$ of node $i$ in hidden layer $t$ is defined as:
E(3)不变性的保持 $\pmb{}$ E(3)等变性。上述操作能够保持输入原子特征与几何信息的E(3)不变性,并可预测E(3)不变的标量属性。为预测E(3)等变的矢量属性 $\vec{u}$ ,我们为每个节点 $i$ 引入关联几何向量 $\vec{\nu}{i}$ ,并扩展PAMNet以保持学习 $\vec{u}$ 时的E(3)等变性。具体而言,隐藏层 $t$ 中节点 $i$ 的关联几何向量 $\vec{\nu}_{i}^{t}$ 定义为:
$$
\vec{\nu}{i}^{t}=f_{\nu}({h^{t}},{\vec{r}}),
$$
$$
\vec{\nu}{i}^{t}=f_{\nu}({h^{t}},{\vec{r}}),
$$
where ${h^{t}}$ denotes the set of learned node embeddings of all nodes in hidden layer $t.$ , ${{\vec{r}}}$ denotes the set of position vectors of all nodes in 3d coordinate space, and $f_{\nu}$ is a function that preserves the E(3)-e qui variance of $\vec{\nu}_{i}^{t}$ with respect to ${{\vec{r}}}$ . Equation (8) is computed after each message passing module in PAMNet.
其中 ${h^{t}}$ 表示隐藏层 $t$ 中所有节点的学习节点嵌入集合,${{\vec{r}}}$ 表示三维坐标空间中所有节点的位置向量集合,$f_{\nu}$ 是一个保持 $\vec{\nu}_{i}^{t}$ 相对于 ${{\vec{r}}}$ 的 E(3) 等变性的函数。PAMNet 中每个消息传递模块后都会计算方程 (8)。
To predict a final vectorial property $\vec{u}$ , we modify Equation (6) and (7) in the fusion module as the following operations:
为了预测最终的矢量属性 $\vec{u}$,我们在融合模块中修改了方程(6)和(7),具体操作如下:
$$
\begin{array}{r l}&{\vec{u}{i}^{t}=\sum_{p}\alpha_{p,i}^{t}(\boldsymbol{W}{p}^{'t}\mathbf{z}{p,i}^{t})\vec{\nu}{p,i}^{t},}\ &{\vec{u}=\sum_{i=1}^{N}\sum_{t=1}^{T}\vec{u}_{i}^{t},}\end{array}
$$
$$
\begin{array}{r l}&{\vec{u}{i}^{t}=\sum_{p}\alpha_{p,i}^{t}(\boldsymbol{W}{p}^{'t}\mathbf{z}{p,i}^{t})\vec{\nu}{p,i}^{t},}\ &{\vec{u}=\sum_{i=1}^{N}\sum_{t=1}^{T}\vec{u}_{i}^{t},}\end{array}
$$
where $\vec{\nu}{p,i}^{t}$ is the associated geometric vector of node $i$ on graph layer $p$ in hidden layer $t$ , $\vec{u}{i}^{t}$ is the learned vector of node $i$ in hidden layer $t$ , and $W_{p}^{'t}\in\mathbb{R}^{1\times F}$ is a learnable weight matrix different for each hidden layer $t$ and graph layer $p$ . In Equation (9), we multiply $\vec{\nu}_{p,i}^{t}$ with the learned scalar node contributions. In Equation (10), we sum all node-level vectors in all hidden layers to compute the final prediction $\vec{u}$ .
其中 $\vec{\nu}{p,i}^{t}$ 是隐藏层 $t$ 中图层级 $p$ 上节点 $i$ 的关联几何向量,$\vec{u}{i}^{t}$ 是隐藏层 $t$ 中节点 $i$ 的学习向量,$W_{p}^{'t}\in\mathbb{R}^{1\times F}$ 是可学习的权重矩阵,每个隐藏层 $t$ 和图层级 $p$ 都不同。在公式 (9) 中,我们将 $\vec{\nu}_{p,i}^{t}$ 与学习到的标量节点贡献相乘。在公式 (10) 中,我们对所有隐藏层中的节点级向量求和以计算最终预测 $\vec{u}$。
where $\left\Vert\cdot\right\Vert$ denotes the L2 norm. Since $\vec{\nu}_{i}^{t}$ as well as $\vec{\mu}$ are computed by a linear combination of ${{\vec{r}}}$ , our PAMNet can preserve E(3)-e qui variance with respect to ${{\vec{r}}}$ when performing the prediction.
其中 $\left\Vert\cdot\right\Vert$ 表示 L2 范数。由于 $\vec{\nu}_{i}^{t}$ 和 $\vec{\mu}$ 都是通过 ${{\vec{r}}}$ 的线性组合计算得出,我们的 PAMNet 在进行预测时能够保持关于 ${{\vec{r}}}$ 的 E(3) 等变性。
Computational Complexity. We analyze the computational complexity of PAMNet by addressing the number of messages. We denote the cutoff distance when creating the edges as $d_{g}$ and $d_{l}$ in $G_{g}$ and $G_{l}$ . The average degree is $k_{g}$ in $G_{g}$ and is $k_{l}$ in $G_{l}$ . In each hidden layer of PAMNet, Global Message Passing needs $O(N k_{g})$ messages because it requires one message for each pairwise distance between the central node and its one-hop neighbor. While Local Message Passing requires one message for each one-hop or two-hop angle around the central node. The number of angles can be estimated as follows: For $k$ edges connected to a node, they can define $(k(k-1))/2$ angles which result in a complexity of $O(N k^{2})$ . The number of one-hop angles and two-hop angles all has such complexity. So that Local Message Passing needs ${\cal O}(2N{k_{l}}^{2})$ messages. In total, PAMNet requires the computation of $O(N k_{g}+2N k_{l}^{~2})$ messages in each hidden layer, while previous approaches 15–18,27 require ${\cal O}(N{k_{g}}^{2})$ messages. For 3D molecules, we have $k_{g}\propto{d_{g}}^{3}$ and $k_{l}\propto d_{l}^{3}$ . With proper choices of $d_{l}$ and $d_{g}$ , we have $k_{l}\ll k_{g}$ . In such cases, our model is more efficient than the related GNNs. We here list the comparison of the number of messages needed in our experiments as an example: On QM9 with , our model needs $0.5\mathrm{k}$ messages/molecule on average, while DimeNet+ $^+$ needs $4.3\mathrm{k}$ messages. On PDBBind with , our model needs only $12\mathrm{k\Omega}$ messages/molecule on average, while DimeNet++ needs 264k messages.
计算复杂度。我们通过分析消息数量来评估PAMNet的计算复杂度。设构建边时的截断距离在全局图$G_{g}$和局部图$G_{l}$中分别为$d_{g}$和$d_{l}$,平均度数在$G_{g}$中为$k_{g}$,在$G_{l}$中为$k_{l}$。PAMNet每个隐藏层中,全局消息传递需要$O(N k_{g})$次消息计算(中心节点与一阶邻居的每对距离需传递一次消息),而局部消息传递需为中心节点周围每个一阶或二阶角度传递消息。角度数量可估算为:$k$条边可定义$(k(k-1))/2$个角度,复杂度为$O(N k^{2})$。一阶与二阶角度均具有该复杂度,故局部消息传递需${\cal O}(2N{k_{l}}^{2})$次消息。总体而言,PAMNet每隐藏层需计算$O(N k_{g}+2N k_{l}^{~2})$次消息,而先前方法[15-18,27]需要${\cal O}(N{k_{g}}^{2})$次消息。对于3D分子体系,存在$k_{g}\propto{d_{g}}^{3}$和$k_{l}\propto d_{l}^{3}$的关系。当合理选择$d_{l}$与$d_{g}$时,可满足$k_{l}\ll k_{g}$,此时本模型比相关图神经网络更高效。以实验数据为例:在QM9数据集上,本模型平均需$0.5\mathrm{k}$次消息/分子,而DimeNet$^+$需$4.3\mathrm{k}$次;在PDBBind数据集上,本模型仅需$12\mathrm{k\Omega}$次消息/分子,DimeNet++则需264k次。
Data collection and processing
数据收集与处理
QM9. For QM9, we use the source provided by24. Following the previous works15–17, we process QM9 by removing about 3k molecules that fail a geometric consistency check or are difficult to converge45. For properties $U_{0},U,H$ , and $G$ , only the a to miz ation energies are used by subtracting the atomic reference energies as $\mathrm{in^{15-17}}$ . For property $\Delta\varepsilon$ , we follow the same way as the DFT calculation and predict it by calculating $\varepsilon_{\mathrm{LUMO}}-\varepsilon_{\mathrm{HOMO}}$ . For property $\mu$ , the final result is the magnitude of the predicted vectorial $\mu$ when using our geometric vector-based approaches with PAMNet. The 3D molecular structures are processed using the RDKit library46. Following 15, we randomly use 110000 molecules for training, 10000 for validation and 10831 for testing. In our multiplex molecular graphs, we use chemical bonds as the edges in the local layer, and a cutoff distance (5 or $10\mathrm{\mathring{A}}.$ ) to create the edges in the global layer.
QM9. 对于QM9数据集,我们采用文献[24]提供的原始数据。遵循先前研究[15-17]的方法,我们通过剔除约3000个未能通过几何一致性检查或难以收敛[45]的分子来处理QM9数据。对于属性$U_{0},U,H$和$G$,仅使用通过减去原子参考能量得到的异构化能,如文献[15-17]所示。对于属性$\Delta\varepsilon$,我们采用与DFT计算相同的方式,通过计算$\varepsilon_{\mathrm{LUMO}}-\varepsilon_{\mathrm{HOMO}}$进行预测。对于属性$\mu$,当使用我们基于几何向量的PAMNet方法时,最终结果是预测矢量$\mu$的模长。三维分子结构使用RDKit库[46]进行处理。按照文献[15]的方法,我们随机选取110000个分子用于训练,10000个用于验证,10831个用于测试。在我们的多重分子图中,局部层使用化学键作为边,全局层则采用截断距离(5或$10\mathrm{\mathring{A}}$)来构建边。
RNA-Puzzles. RNA-Puzzles consists of the first 21 RNAs in the RNA-Puzzles structure prediction challenge 32. Each RNA is used to generate at least 1500 structural models using FARFAR2, where $1%$ of the models are near native (i.e., within a $2\mathring\mathrm{A}$ RMSD of the experimentally determined native structure). Following 30, we only use the carbon, nitrogen, and oxygen atoms in RNA structures. When building multiplex graphs for RNA structures, we use cutoff distance $d_{l}=2.6\mathring\mathrm{A}$ for the local interactions in $G_{l o c a l}$ and $d_{g}=20\mathring\mathrm{A}$ for the global interactions in $G_{g l o b a l}$ .
RNA-Puzzles。RNA-Puzzles包含RNA-Puzzles结构预测挑战32中的前21个RNA。每个RNA使用FARFAR2生成至少1500个结构模型,其中$1%$的模型接近天然结构(即与实验确定的天然结构的RMSD在$2\mathring\mathrm{A}$以内)。根据[30],我们仅使用RNA结构中的碳、氮和氧原子。在构建RNA结构的多路图时,局部相互作用$G_{local}$的截止距离$d_{l}=2.6\mathring\mathrm{A}$,全局相互作用$G_{global}$的截止距离$d_{g}=20\mathring\mathrm{A}$。
PDBBind. For PDBBind, we use PDBbind v2016 following 18,36. Besides, we use the same data splitting method according to18 for a fair comparison. In detail, we use the core subset which contains 290 complexes in PDBbind v2016 for testing. The difference between the refined and core subsets, which includes 3767 complexes, is split with a ratio of 9:1 for training and validation. We use $\mathrm{log}K_{i}$ as the target property being predicted, which is proportional to the binding free energy. In each complex, we exclude the protein residues that are more than from the ligand and remove all hydrogen atoms. The resulting complexes contain around 300 atoms on average. In our multiplex molecular graphs, we use cutoff distance in the local layer and in the global layer.
PDBBind。对于PDBBind,我们遵循[18,36]使用PDBbind v2016版本。此外,为公平比较,我们采用与[18]相同的数据划分方法。具体而言,我们使用PDBbind v2016中包含290个复合物的核心子集进行测试。精炼子集与核心子集之间的差异(包含3767个复合物)按9:1的比例划分为训练集和验证集。我们以$\mathrm{log}K_{i}$(与结合自由能成正比)作为预测目标属性。在每个复合物中,我们排除距离配体超过的蛋白质残基,并移除所有氢原子。最终得到的复合物平均包含约300个原子。在我们的多重分子图中,局部层使用截断距离,全局层使用。
Experimental settings
实验设置
In our message passing operations, we define $\phi_{d}(e)=W_{e}e$ and $\phi_{\alpha}(\alpha)=\mathrm{MLP}_{\alpha}(\alpha)$ , where $W_{e}$ is a weight matrix, $\mathrm{MLP}_{\alpha}$ is a multi-layer perceptron (MLP). All MLPs used in our model have two layers by taking advantage of the approximation capability of $\mathrm{MLP^{47}}$ . For all activation functions, we use the self-gated Swish activation function48. For the basis functions, we use the same parameters as $\mathrm{in}^{15}$ . To initialize all learnable parameters, we use the default settings used in PyTorch without assigning specific initialization s except the initialization for the input node embeddings on QM9: $h$ are initialized with random values uniformly distributed between $-\sqrt{3}$ and $\sqrt{3}$ . In all experiments, we use the Adam optimizer 49 to minimize the loss. In Supplementary Table S3, we list the typical hyper parameters used in our experiments. All of the experiments are done on an NVIDIA Tesla V100 GPU (32 GB).
在我们的消息传递操作中,定义 $\phi_{d}(e)=W_{e}e$ 和 $\phi_{\alpha}(\alpha)=\mathrm{MLP}_{\alpha}(\alpha)$ ,其中 $W_{e}$ 是权重矩阵, $\mathrm{MLP}_{\alpha}$ 为多层感知机 (MLP) 。模型中的所有MLP均采用两层结构,以利用 $\mathrm{MLP^{47}}$ 的近似能力。所有激活函数均采用自门控Swish激活函数48。基函数参数设置与 $\mathrm{in}^{15}$ 相同。可学习参数初始化除QM9数据集输入节点嵌入 $h$ 采用 $-\sqrt{3}$ 至 $\sqrt{3}$ 均匀分布的随机值外,其余均采用PyTorch默认设置。所有实验使用Adam优化器49进行损失最小化,典型超参数列于补充表S3。实验均在NVIDIA Tesla V100 GPU (32 GB) 上完成。
Small molecule property prediction. In our experiment on QM9, we use the single-target training following 15 by using a separate model for each target instead of training a single shared model for all targets. The models are optimized by minimizing the mean absolute error (MAE) loss. We use a linear learning rate warm-up over 1 epoch and an exponential decay with a ratio 0.1 every 600 epochs. The model parameter values for validation and testing are kept using an exponential moving average with a decay rate of 0.999. To prevent over fitting, we use early stopping on the validation loss. For properties ZPVE, $U_{0},U,H$ , and $G$ , we use the cutoff distance in the global layer . For the other properties, we use $ . We repeat our runs 3 times for each PAMNet variant following 50.
小分子性质预测。在QM9数据集上的实验中,我们采用单目标训练策略[15],即针对每个预测目标使用独立模型而非共享模型。模型通过最小化平均绝对误差(MAE)损失进行优化,采用线性学习率预热(持续1个epoch)和指数衰减(每600个epoch衰减比例为0.1)。验证集和测试集的模型参数采用指数移动平均保存(衰减率0.999)。为防止过拟合,我们根据验证集损失进行早停。对于ZPVE、$U_{0},U,H$和$G$性质,全局层截断距离设为;其余性质则使用。每种PAMNet变体均重复实验3次[50]。
RNA 3D structure prediction. PAMNet is optimized by minimizing the smooth L1 loss51 between the predicted value and the ground truth. An early-stopping strategy is adopted to decide the best epoch based on the validation loss.
RNA 3D结构预测。PAMNet通过最小化预测值与真实值之间的平滑L1损失51进行优化,并采用早停策略根据验证损失确定最佳训练轮次。
Protein-ligand binding affinity prediction. We create three weight-sharing, replica networks, one each for predicting the target $G$ of complex, protein pocket, and ligand following 52. The final target is computed by $\Delta G_{\mathrm{complex}}=G_{\mathrm{complex}}-G_{\mathrm{pocket}}-$ $G_{\mathrm{ligand}}$ . The full model is trained by minimizing the mean absolute error (MAE) loss between $\Delta G_{\mathrm{complex}}$ and the true values. The learning rate is dropped by a factor of 0.2 every 50 epochs. Moreover, we perform 5 independent runs according to18.
蛋白质-配体结合亲和力预测。我们创建了三个权重共享的复制网络,分别用于预测复合物、蛋白质口袋和配体的目标 $G$,遵循[52]。最终目标通过 $\Delta G_{\mathrm{complex}}=G_{\mathrm{complex}}-G_{\mathrm{pocket}}-$ $G_{\mathrm{ligand}}$ 计算得出。完整模型通过最小化 $\Delta G_{\mathrm{complex}}$ 与真实值之间的平均绝对误差(MAE)损失进行训练。学习率每50个周期下降0.2倍。此外,我们根据[18]进行了5次独立运行。
Efficiency comparison. In the experiment on investigating the efficiency of PAMNet, we use NVIDIA Tesla V100 GPU (32 GB) for a fair comparison. For small molecule property prediction, we use the related models for predicting property $U_{0}$ of QM9 as an example. We use batch size ${:=128}$ for all models and use the configurations reported in the corresponding papers. For RNA 3D structure prediction, we use PAMNet and ARES to predict the structural models of RNA in puzzle 5 of RNA-Puzzles challenge. The RNA being predicted has 6034 non-hydrogen atoms. The model settings of PAMNet and ARES are the same as those used for reproducing the best results. We use batch size $_{:=8}$ when performing the predictions. For protein-ligand binding affinity prediction, we use the configurations that can reproduce the best results for the related models.
效率对比。在评估PAMNet效率的实验中,我们使用NVIDIA Tesla V100 GPU(32 GB)以确保公平比较。针对小分子性质预测,我们以QM9数据集的性质$U_{0}$预测为例,所有模型均采用批次大小${:=128}$,并沿用原论文报告的配置。对于RNA三维结构预测,我们采用PAMNet和ARES预测RNA-Puzzles挑战赛第五题中的RNA结构模型,目标RNA包含6034个非氢原子。PAMNet与ARES的模型设置与其最佳结果复现时保持一致,预测时采用批次大小$_{:=8}$。在蛋白质-配体结合亲和力预测任务中,所有相关模型均采用可复现最佳结果的配置参数。
Data Availability
数据可用性
The QM9 dataset is available at https://figshare.com/collections/Quantum chemistry structures and properties of 134 kilo molecules/978904. The datasets for RNA 3D structure prediction can be found at https://purl.stanford.edu/bn 398 fc 4306. The PDBbind v2016 dataset is available at http://www.pdbbind.org.cn/ or https://github.com/Paddle Paddle/Paddle Helix/tree/dev/ apps/drug target interaction/sign.
QM9数据集可在https://figshare.com/collections/Quantum chemistry structures and properties of 134 kilo molecules/978904获取。RNA三维结构预测数据集位于https://purl.stanford.edu/bn 398 fc 4306。PDBbind v2016数据集可通过http://www.pdbbind.org.cn/或https://github.com/Paddle Paddle/Paddle Helix/tree/dev/ apps/drug target interaction/sign下载。
Code Availability
代码可用性
The source code of our model is publicly available on GitHub at the following repository: https://github.com/Xie Research Group Physics-aware-Multiplex-GNN.
我们模型的源代码已在GitHub上公开,仓库地址为:https://github.com/Xie Research Group Physics-aware-Multiplex-GNN。