BERTOLOGY MEETS BIOLOGY: INTERPRETING ATTENTION IN PROTEIN LANGUAGE MODELS
BERTOLOGY 遇见生物学:解读蛋白质语言模型中的注意力机制
ABSTRACT
摘要
Transformer architectures have proven to learn useful representations for protein classification and generation tasks. However, these representations present challenges in interpret ability. In this work, we demonstrate a set of methods for analyzing protein Transformer models through the lens of attention. We show that attention: (1) captures the folding structure of proteins, connecting amino acids that are far apart in the underlying sequence, but spatially close in the three-dimensional structure, (2) targets binding sites, a key functional component of proteins, and (3) focuses on progressively more complex bio physical properties with increasing layer depth. We find this behavior to be consistent across three Transformer architectures (BERT, ALBERT, XLNet) and two distinct protein datasets. We also present a three-dimensional visualization of the interaction between attention and protein structure. Code for visualization and analysis is available at https://github.com/salesforce/provis.
Transformer架构已被证明能有效学习蛋白质分类和生成任务的有用表征。然而,这些表征在可解释性方面存在挑战。本工作展示了一套通过注意力机制分析蛋白质Transformer模型的方法。我们发现注意力机制能够:(1) 捕捉蛋白质的折叠结构,连接底层序列中相距较远但在三维结构中空间邻近的氨基酸,(2) 靶向蛋白质关键功能组件——结合位点,(3) 随着网络层深的增加,逐步聚焦更复杂的生物物理特性。该行为在三种Transformer架构(BERT、ALBERT、XLNet)和两个不同蛋白质数据集中表现一致。我们还提供了注意力与蛋白质结构交互的三维可视化。可视化与分析代码详见https://github.com/salesforce/provis。
1 INTRODUCTION
1 引言
The study of proteins, the fundamental macromolecules governing biology and life itself, has led to remarkable advances in understanding human health and the development of disease therapies. The decreasing cost of sequencing technology has enabled vast databases of naturally occurring proteins (El-Gebali et al., 2019a), which are rich in information for developing powerful machine learning models of protein sequences. For example, sequence models leveraging principles of co-evolution, whether modeling pairwise or higher-order interactions, have enabled prediction of structure or function (Rollins et al., 2019).
蛋白质研究作为理解生物体运作机制和生命本质的基础大分子科学,已推动人类健康认知与疾病治疗领域的重大突破。随着测序技术成本持续下降,天然蛋白质数据库规模不断扩大[20],这些海量数据为开发强大的蛋白质序列机器学习模型提供了丰富素材。例如基于共进化原理的序列模型(无论建模双元或高阶相互作用)已能有效预测蛋白质结构或功能[21]。
Proteins, as a sequence of amino acids, can be viewed precisely as a language and therefore modeled using neural architectures developed for natural language. In particular, the Transformer (Vaswani et al., 2017), which has revolutionized unsupervised learning for text, shows promise for similar impact on protein sequence modeling. However, the strong performance of the Transformer comes at the cost of interpret ability, and this lack of transparency can hide underlying problems such as model bias and spurious correlations (Niven & Kao, 2019; Tan & Celis, 2019; Kurita et al., 2019). In response, much NLP research now focuses on interpreting the Transformer, e.g., the sub specialty of “BERTology” (Rogers et al., 2020), which specifically studies the BERT model (Devlin et al., 2019).
蛋白质作为氨基酸序列,可被精确视为一种语言,因此能够用为自然语言开发的神经架构进行建模。特别是Transformer (Vaswani等人,2017) 这一彻底改变文本无监督学习的技术,有望在蛋白质序列建模领域产生类似影响。但Transformer的强大性能是以可解释性为代价的,这种透明度的缺失可能掩盖模型偏差和伪相关等潜在问题 (Niven & Kao, 2019; Tan & Celis, 2019; Kurita等人,2019) 。对此,当前大量自然语言处理研究聚焦于Transformer的可解释性,例如专门研究BERT模型 (Devlin等人,2019) 的子领域"BERTology" (Rogers等人,2020) 。
In this work, we adapt and extend this line of interpret ability research to protein sequences. We analyze Transformer protein models through the lens of attention, and present a set of interpret ability methods that capture the unique functional and structural characteristics of proteins. We also compare the knowledge encoded in attention weights to that captured by hidden-state representations. Finally, we present a visualization of attention contextual i zed within three-dimensional protein structure.
在本工作中,我们调整并扩展了这类可解释性研究以适用于蛋白质序列。我们通过注意力机制(attention)的视角分析Transformer蛋白质模型,提出了一套能捕捉蛋白质独特功能与结构特征的可解释性方法。同时,我们将注意力权重(attention weights)编码的知识与隐藏状态表示(hidden-state representations)捕获的知识进行对比。最后,我们展示了在三维蛋白质结构语境下的注意力可视化结果。
Our analysis reveals that attention captures high-level structural properties of proteins, connecting amino acids that are spatially close in three-dimensional structure, but apart in the underlying sequence (Figure 1a). We also find that attention targets binding sites, a key functional component of proteins (Figure 1b). Further, we show how attention is consistent with a classic measure of similarity between amino acids—the substitution matrix. Finally, we demonstrate that attention captures progressively higher-level representations of structure and function with increasing layer depth.
我们的分析表明,注意力机制能够捕捉蛋白质的高级结构特性,将三维结构中空间距离相近但底层序列上相隔较远的氨基酸连接起来(图 1a)。我们还发现,注意力机制会聚焦于结合位点——蛋白质的关键功能组成部分(图 1b)。此外,我们展示了注意力机制如何与经典的氨基酸相似性度量标准——替换矩阵保持一致。最后,我们证明随着网络层深的增加,注意力机制能逐步捕获更高层次的结构与功能表征。
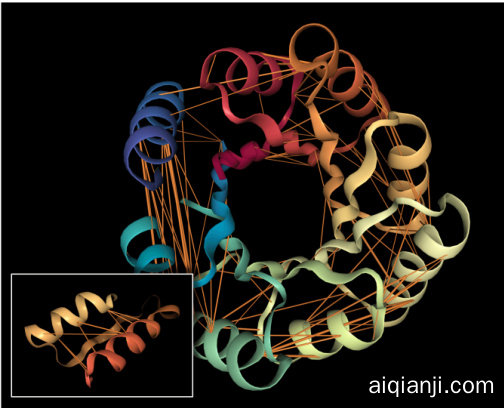
(a) Attention in head 12-4, which targets amino acid pairs that are close in physical space (see inset sub sequence 117D-157I) but lie apart in the sequence. Example is a de novo designed TIMbarrel (5BVL) with characteristic symmetry.
(a) 头12-4中的注意力机制,其针对物理空间邻近但序列上相距较远的氨基酸对(见插图中子序列117D-157I)。示例为一个具有特征对称性的从头设计TIM桶状结构(5BVL)。
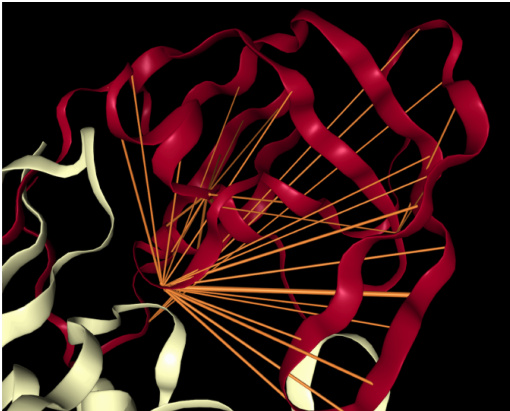
(b) Attention in head 7-1, which targets binding sites, a key functional component of proteins. Example is HIV-1 protease (7HVP). The primary location receiving attention is 27G, a binding site for protease inhibitor small-molecule drugs.
图 1:
(b) 头 7-1 中的注意力机制,其靶向蛋白质的关键功能组件——结合位点。示例为 HIV-1 蛋白酶 (7HVP)。主要接收注意力的位置是 27G,这是蛋白酶抑制剂小分子药物的结合位点。
Figure 1: Examples of how specialized attention heads in a Transformer recover protein structure and function, based solely on language model pre-training. Orange lines depict attention between amino acids (line width proportional to attention weight; values below 0.1 hidden). Heads were selected based on correlation with ground-truth annotations of contact maps and binding sites. Visualization s based on the NGL Viewer (Rose et al., 2018; Rose & Hildebrand, 2015; Nguyen et al., 2017).
图 1: Transformer中专一注意力头仅通过语言模型预训练恢复蛋白质结构和功能的示例。橙色线条表示氨基酸之间的注意力(线条宽度与注意力权重成正比;低于0.1的值已隐藏)。注意力头的选择基于与接触图和结合位点真实标注的相关性。可视化基于NGL Viewer (Rose et al., 2018; Rose & Hildebrand, 2015; Nguyen et al., 2017)。
In contrast to NLP, which aims to automate a capability that humans already have—understanding natural language—protein modeling also seeks to shed light on biological processes that are not fully understood. Thus we also discuss how interpret ability can aid scientific discovery.
与旨在自动化人类已有能力(理解自然语言)的 NLP 不同,蛋白质建模还试图揭示尚未完全理解的生物过程。因此,我们也讨论了可解释性如何助力科学发现。
2 BACKGROUND: PROTEINS
2 背景:蛋白质
In this section we provide background on the biological concepts discussed in later sections.
在本节中,我们将为后续章节讨论的生物学概念提供背景知识。
Amino acids. Just as language is composed of words from a shared lexicon, every protein sequence is formed from a vocabulary of amino acids, of which 20 are commonly observed. Amino acids may be denoted by their full name (e.g., Proline), a 3-letter abbreviation $(P r o)$ , or a single-letter code $(P)$
氨基酸。正如语言由共享词汇表中的单词组成一样,每个蛋白质序列都是由氨基酸词汇构成的,其中常见的有20种。氨基酸可以用全名(如Proline)、3字母缩写$(Pro)$或单字母代码$(P)$表示。
Substitution matrix. While word synonyms are encoded in a thesaurus, proteins that are similar in structure or function are captured in a substitution matrix, which scores pairs of amino acids on how readily they may be substituted for one another while maintaining protein viability. One common substitution matrix is BLOSUM (Henikoff & Henikoff, 1992), which is derived from co-occurrence statistics of amino acids in aligned protein sequences.
替换矩阵 (Substitution matrix)。虽然单词同义词被编码在同义词词典中,但结构或功能相似的蛋白质则通过替换矩阵来捕捉,该矩阵通过评分氨基酸对的替换可能性来评估它们在维持蛋白质活性时的可替代性。常见的替换矩阵是 BLOSUM (Henikoff & Henikoff, 1992),它源自对齐蛋白质序列中氨基酸的共现统计。
Protein structure. Though a protein may be abstracted as a sequence of amino acids, it represents a physical entity with a well-defined three-dimensional structure (Figure 1). Secondary structure describes the local segments of proteins; two commonly observed types are the alpha helix and beta sheet. Tertiary structure encompasses the large-scale formations that determine the overall shape and function of the protein. One way to characterize tertiary structure is by a contact map, which describes the pairs of amino acids that are in contact (within 8 angstroms of one another) in the folded protein structure but lie apart (by at least 6 positions) in the underlying sequence (Rao et al., 2019).
蛋白质结构。尽管蛋白质可以被抽象为氨基酸序列,但它代表具有明确三维结构的物理实体(图 1)。二级结构描述了蛋白质的局部片段;两种常见类型是α螺旋和β折叠。三级结构包含决定蛋白质整体形状和功能的大规模构象。表征三级结构的一种方法是通过接触图(contact map),它描述了在折叠蛋白质结构中相互接触(彼此距离在8埃以内)但在基础序列中相隔较远(至少相隔6个位置)的氨基酸对(Rao et al., 2019)。
Binding sites. Proteins may also be characterized by their functional properties. Binding sites are protein regions that bind with other molecules (proteins, natural ligands, and small-molecule drugs) to carry out a specific function. For example, the HIV-1 protease is an enzyme responsible for a critical process in replication of HIV (Brik & Wong, 2003). It has a binding site, shown in Figure 1b, that is a target for drug development to ensure inhibition.
结合位点。蛋白质也可以通过其功能特性来描述。结合位点是蛋白质中与其他分子(蛋白质、天然配体和小分子药物)结合以执行特定功能的区域。例如,HIV-1蛋白酶是一种在HIV复制过程中起关键作用的酶(Brik & Wong, 2003)。如图1b所示,它具有一个结合位点,是药物开发中确保抑制作用的靶点。
Post-translational modifications. After a protein is translated from RNA, it may undergo additional modifications, e.g. phosphor yl ation, which play a key role in protein structure and function.
翻译后修饰。蛋白质从RNA翻译后,可能经历额外的修饰,例如磷酸化 (phosphorylation),这对蛋白质结构和功能起着关键作用。
3 METHODOLOGY
3 方法论
Model. We demonstrate our interpret ability methods on five Transformer models that were pretrained through language modeling of amino acid sequences. We primarily focus on the BERT-Base model from TAPE (Rao et al., 2019), which was pretrained on Pfam, a dataset of 31M protein sequences (ElGebali et al., 2019b). We refer to this model as TapeBert. We also analyze 4 pre-trained Transformer models from ProtTrans (Elnaggar et al., 2020): ProtBert and ProtBert-BFD, which are 30-layer, 16-head BERT models; ProtAlbert, a 12-layer, 64-head ALBERT (Lan et al., 2020) model; and ProtXLNet, a 30-layer, 16-head XLNet (Yang et al., 2019) model. ProtBert-BFD was pretrained on BFD (Steinegger & Söding, 2018), a dataset of 2.1B protein sequences, while the other ProtTrans models were pretrained on UniRef100 (Suzek et al., 2014), which includes 216M protein sequences. A summary of these 5 models is presented in Appendix A.1.
模型。我们在五个通过氨基酸序列语言建模预训练的Transformer模型上展示了可解释性方法。主要研究对象是TAPE (Rao等人,2019) 中的BERT-Base模型,该模型在包含3100万条蛋白质序列的Pfam数据集 (ElGebali等人,2019b) 上完成预训练,我们称其为TapeBert。同时分析了ProtTrans (Elnaggar等人,2020) 的四个预训练Transformer模型:ProtBert和ProtBert-BFD(30层16头BERT架构)、ProtAlbert(12层64头ALBERT架构 (Lan等人,2020)),以及ProtXLNet(30层16头XLNet架构 (Yang等人,2019))。其中ProtBert-BFD基于21亿条序列的BFD数据集 (Steinegger & Söding,2018) 预训练,其余ProtTrans模型则在包含2.16亿条序列的UniRef100 (Suzek等人,2014) 上训练。这五个模型的详细参数见附录A.1。
Here we present an overview of BERT, with additional details on all models in Appendix A.2. BERT inputs a sequence of amino acids $\pmb{x}=(x_ {1},\dots,x_ {n})$ and applies a series of encoders. Each encoder layer $\ell$ outputs a sequence of continuous embeddings $(\mathbf{h}_ {1}^{(\ell)},\dots,\mathbf{h}_ {n}^{(\ell)})$ using a multi-headed attention mechanism. Each attention head in a layer produces a set of attention weights $\alpha$ for an input, where $\alpha_ {i,j}>0$ is the attention from token $i$ to token $j$ , such that $\textstyle\sum_ {j}\alpha_ {i,j}=1$ . Intuitively, attention weights define the influence of every token on the next layer’s repr esentation for the current token. We denote a particular head by
这里我们概述BERT,更多关于所有模型的细节见附录A.2。BERT输入一个氨基酸序列$\pmb{x}=(x_ {1},\dots,x_ {n})$,并应用一系列编码器。每个编码器层$\ell$通过多头注意力机制输出一个连续嵌入序列$(\mathbf{h}_ {1}^{(\ell)},\dots,\mathbf{h}_ {n}^{(\ell)})$。每层的注意力头为输入生成一组注意力权重$\alpha$,其中$\alpha_ {i,j}>0$表示从token $i$到token $j$的注意力,满足$\textstyle\sum_ {j}\alpha_ {i,j}=1$。直观上,注意力权重定义了每个token对当前token下一层表示的影响。我们用<层号>-<头索引>表示特定头,例如3-7表示第3层的第7个头。
Attention analysis. We analyze how attention aligns with various protein properties. For properties of token pairs, e.g. contact maps, we define an indicator function $f(i,j)$ that returns 1 if the property is present in token pair $(i,j)$ (e.g., if amino acids $i$ and $j$ are in contact), and 0 otherwise. We then compute the proportion of high-attention token pairs $(\alpha_ {i,j}>\theta)$ ) where the property is present, aggregated over a dataset $\boldsymbol{X}$ :
注意力分析。我们分析注意力如何与各种蛋白质特性对齐。对于token对的特性,例如接触图,我们定义一个指示函数$f(i,j)$,如果特性存在于token对$(i,j)$中(例如,氨基酸$i$和$j$接触),则返回1,否则返回0。然后我们计算高注意力token对$(\alpha_ {i,j}>\theta)$中存在该特性的比例,在数据集$\boldsymbol{X}$上聚合:
$$
p_ {\alpha}(f)=\sum_ {\mathbf{x}\in\mathbf{X}}\sum_ {i=1}^{|\mathbf{x}|}\sum_ {j=1}^{|\mathbf{x}|}f(i,j)\cdot\mathbb{1}_ {\alpha_ {i,j}>\theta}{\bigg/}\sum_ {\mathbf{x}\in\mathbf{X}}\sum_ {i=1}^{|\mathbf{x}|}\sum_ {j=1}^{|\mathbf{x}|}\mathbb{1}_ {\alpha_ {i,j}>\theta}
$$
$$
p_ {\alpha}(f)=\sum_ {\mathbf{x}\in\mathbf{X}}\sum_ {i=1}^{|\mathbf{x}|}\sum_ {j=1}^{|\mathbf{x}|}f(i,j)\cdot\mathbb{1}_ {\alpha_ {i,j}>\theta}{\bigg/}\sum_ {\mathbf{x}\in\mathbf{X}}\sum_ {i=1}^{|\mathbf{x}|}\sum_ {j=1}^{|\mathbf{x}|}\mathbb{1}_ {\alpha_ {i,j}>\theta}
$$
where $\theta$ is a threshold to select for high-confidence attention weights. We also present an alternative, continuous version of this metric in Appendix B.1.
其中 $\theta$ 是用于选择高置信度注意力权重的阈值。我们还在附录 B.1 中提供了该指标的另一种连续版本。
For properties of individual tokens, e.g. binding sites, we define $f(i,j)$ to return 1 if the property is present in token $j$ (e.g. if $j$ is a binding site). In this case, $p_ {\alpha}(f)$ equals the proportion of attention that is directed to the property (e.g. the proportion of attention focused on binding sites).
对于单个token的属性(例如结合位点),我们定义$f(i,j)$在token $j$具有该属性时返回1(例如$j$是结合位点)。此时,$p_ {\alpha}(f)$等于指向该属性的注意力比例(例如聚焦于结合位点的注意力比例)。
When applying these metrics, we include two types of checks to ensure that the results are not due to chance. First, we test that the proportion of attention that aligns with particular properties is significantly higher than the background frequency of these properties, taking into account the Bonferroni correction for multiple hypotheses corresponding to multiple attention heads. Second, we compare the results to a null model, which is an instance of the model with randomly shuffled attention weights. We describe these methods in detail in Appendix B.2.
在应用这些指标时,我们包含了两类检查以确保结果并非偶然。首先,我们测试与特定属性对齐的注意力比例是否显著高于这些属性的背景频率,同时考虑了针对多头注意力对应的多重假设进行Bonferroni校正。其次,我们将结果与零模型(即注意力权重随机打乱的模型实例)进行对比。这些方法的详细说明见附录B.2。
Probing tasks. We also perform probing tasks on the model, which test the knowledge contained in model representations by using them as inputs to a classifier that predicts a property of interest (Veldhoen et al., 2016; Conneau et al., 2018; Adi et al., 2016). The performance of the probing classifier serves as a measure of the knowledge of the property that is encoded in the representation. We run both embedding probes, which assess the knowledge encoded in the output embeddings of each layer, and attention probes (Reif et al., 2019; Clark et al., 2019), which measure the knowledge contained in the attention weights for pairwise features. Details are provided in Appendix B.3.
探测任务。我们还在模型上执行探测任务,通过将模型表征作为分类器的输入来预测目标属性,从而测试这些表征中包含的知识 (Veldhoen et al., 2016; Conneau et al., 2018; Adi et al., 2016)。探测分类器的性能可以衡量表征中编码的该属性知识量。我们既运行了评估各层输出嵌入 (embedding) 中编码知识的嵌入探测,也运行了测量注意力权重中成对特征知识的注意力探测 (Reif et al., 2019; Clark et al., 2019)。详见附录B.3。
Datasets. For our analyses of amino acids and contact maps, we use a curated dataset from TAPE based on ProteinNet (AlQuraishi, 2019; Fox et al., 2013; Berman et al., 2000; Moult et al., 2018), which contains amino acid sequences annotated with spatial coordinates (used for the contact map analysis). For the analysis of secondary structure and binding sites we use the Secondary Structure dataset (Rao et al., 2019; Berman et al., 2000; Moult et al., 2018; Klausen et al., 2019) from TAPE. We employed a taxonomy of secondary structure with three categories: Helix, Strand, and Turn/Bend, with the last two belonging to the higher-level beta sheet category (Sec. 2). We used this taxonomy to study how the model understood structurally distinct regions of beta sheets. We obtained token-level binding site and protein modification labels from the Protein Data Bank (Berman et al., 2000). For analyzing attention, we used a random subset of 5000 sequences from the training split of the respective datasets (note that none of the aforementioned annotations were used in model training). For the diagnostic classifier, we used the respective training splits for training and the validation splits for evaluation. See Appendix B.4 for additional details.
数据集。针对氨基酸和接触图的分析,我们使用了基于ProteinNet (AlQuraishi, 2019; Fox et al., 2013; Berman et al., 2000; Moult et al., 2018) 的TAPE精选数据集,其中包含带有空间坐标注释的氨基酸序列(用于接触图分析)。对于二级结构和结合位点的分析,我们采用了TAPE中的二级结构数据集 (Rao et al., 2019; Berman et al., 2000; Moult et al., 2018; Klausen et al., 2019)。我们采用包含三类二级结构的分类法:螺旋 (Helix)、链 (Strand) 和转角/弯曲 (Turn/Bend),后两者属于更高层次的β折叠类别(见第2节)。我们利用该分类法研究模型如何理解β折叠的结构差异区域。结合位点和蛋白质修饰的token级标签来自蛋白质数据库 (Berman et al., 2000)。注意力分析使用了各数据集训练集中随机抽取的5000条序列子集(注意:上述标注均未用于模型训练)。诊断分类器使用相应训练集进行训练,验证集进行评估。更多细节参见附录B.4。
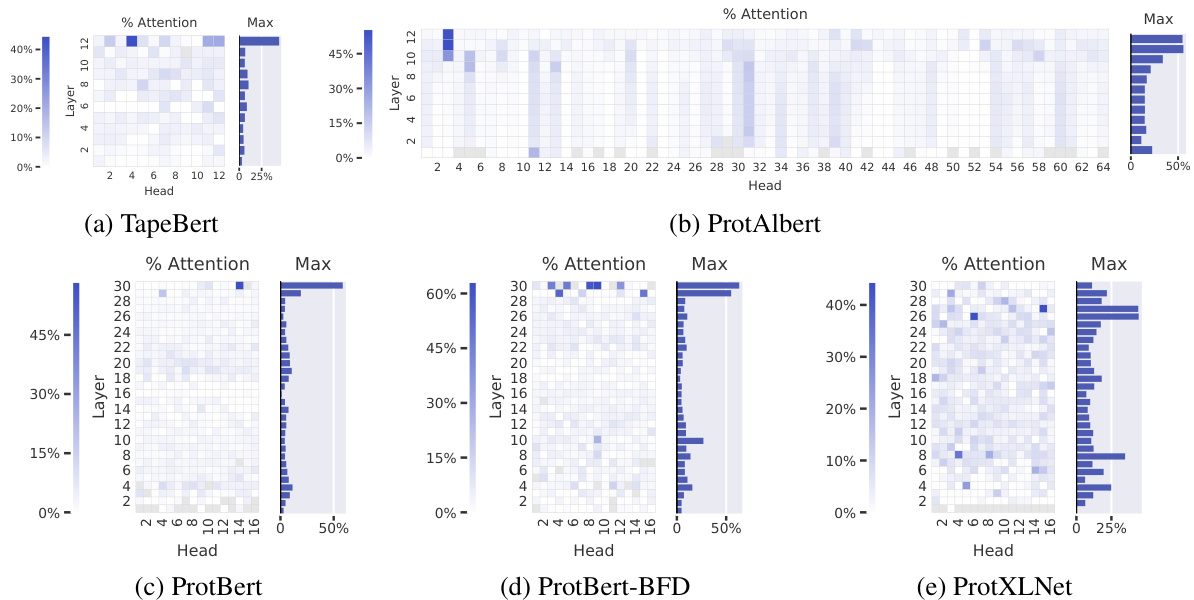
Figure 2: Agreement between attention and contact maps across five pretrained Transformer models from TAPE (a) and ProtTrans (b–e). The heatmaps show the proportion of high-confidence attention weights $(\alpha_ {i,j}>\theta)$ from each head that connects pairs of amino acids that are in contact with one another. In TapeBert (a), for example, we can see that $45%$ of attention in head 12-4 (the 12th layer’s 4th head) maps to contacts. The bar plots show the maximum value from each layer. Note that the vertical striping in ProtAlbert (b) is likely due to cross-layer parameter sharing (see Appendix A.3).
图 2: TAPE (a) 和 ProtTrans (b–e) 五个预训练 Transformer 模型中注意力机制与接触图的一致性关系。热力图展示了每个注意力头中高置信度权重 $(\alpha_ {i,j}>\theta)$ 对应氨基酸接触对的比例。以 TapeBert (a) 为例,可见第12层第4个头 (head 12-4) 中45%的注意力映射到了接触关系上。柱状图显示各层的最大值。需注意 ProtAlbert (b) 中的垂直条纹可能源自跨层参数共享 (参见附录 A.3)。
Experimental details We exclude attention to the [SEP] delimiter token, as it has been shown to be a “no-op” attention token (Clark et al., 2019), as well as attention to the [CLS] token, which is not explicitly used in language modeling. We only include results for attention heads where at least 100 high-confidence attention arcs are available for analysis. We set the attention threshold $\theta$ to 0.3 to select for high-confidence attention while retaining sufficient data for analysis. We truncate all protein sequences to a length of 512 to reduce memory requirements.1
实验细节
我们排除了对[SEP]分隔符token的关注,因为它已被证明是一个"无操作"注意力token (Clark et al., 2019),同时也排除了对[CLS] token的关注,该token在语言建模中并未被显式使用。我们仅包含那些至少有100个高置信度注意力弧可用于分析的注意力头结果。我们将注意力阈值$\theta$设为0.3,以选择高置信度注意力同时保留足够数据进行分析。我们将所有蛋白质序列截断至512长度以减少内存需求。1
We note that all of the above analyses are purely associative and do not attempt to establish a causal link between attention and model behavior (Vig et al., 2020; Grimsley et al., 2020), nor to explain model predictions (Jain & Wallace, 2019; Wiegreffe & Pinter, 2019).
我们注意到,上述所有分析都纯粹是关联性的,并未试图建立注意力与模型行为之间的因果关系 (Vig et al., 2020; Grimsley et al., 2020) ,也未试图解释模型预测 (Jain & Wallace, 2019; Wiegreffe & Pinter, 2019) 。
4 WHAT DOES ATTENTION UNDERSTAND ABOUT PROTEINS?
4 注意力机制对蛋白质的理解是什么?
4.1 PROTEIN STRUCTURE
4.1 蛋白质结构
Here we explore the relationship between attention and tertiary structure, as characterized by contact maps (see Section 2). Secondary structure results are included in Appendix C.1.
我们在此探讨注意力机制与三级结构之间的关系,具体通过接触图 (contact maps) 来表征 (参见第2节)。二级结构相关结果详见附录C.1。
Attention aligns strongly with contact maps in the deepest layers. Figure 2 shows how attention aligns with contact maps across the heads of the five models evaluated , based on the metric defined in Equation 1. The most aligned heads are found in the deepest layers and focus up to $44.7%$ (TapeBert), $55.7%$ (ProtAlbert), $58.5%$ (ProtBert), $63.2%$ (ProtBert-BFD), and $44.5%$ (ProtXLNet) of attention on contacts, whereas the background frequency of contacts among all amino acid pairs in the dataset is $1.3%$ . Figure 1a shows an example of the induced attention from the top head in TapeBert. We note that the model with the single most aligned head—ProtBert-BFD—is the largest model (same size as Protein Bert) at 420M parameters (Appendix A.1) and it was also the only model pre-trained on the largest dataset, BFD. It’s possible that both factors helped the model learn more structurally-aligned attention patterns. Statistical significance tests and null models are reported in Appendix C.2.
注意力在最深层与接触图高度对齐。图 2展示了基于公式1定义的指标,五个评估模型中各注意力头与接触图的对齐情况。对齐度最高的注意力头出现在最深层,其关注接触的比例分别达到:TapeBert (44.7%)、ProtAlbert (55.7%)、ProtBert (58.5%)、ProtBert-BFD (63.2%) 和 ProtXLNet (44.5%),而数据集中所有氨基酸对接触的背景频率仅为1.3%。图1a展示了TapeBert顶层注意力头产生的注意力示例。值得注意的是,具有最高单头对齐度的ProtBert-BFD是参数量最大(4.2亿,与Protein Bert相同)的模型(附录A.1),也是唯一在最大数据集BFD上预训练的模型。这两个因素可能共同促使该模型学习了更具结构对齐性的注意力模式。统计显著性检验与零模型结果见附录C.2。
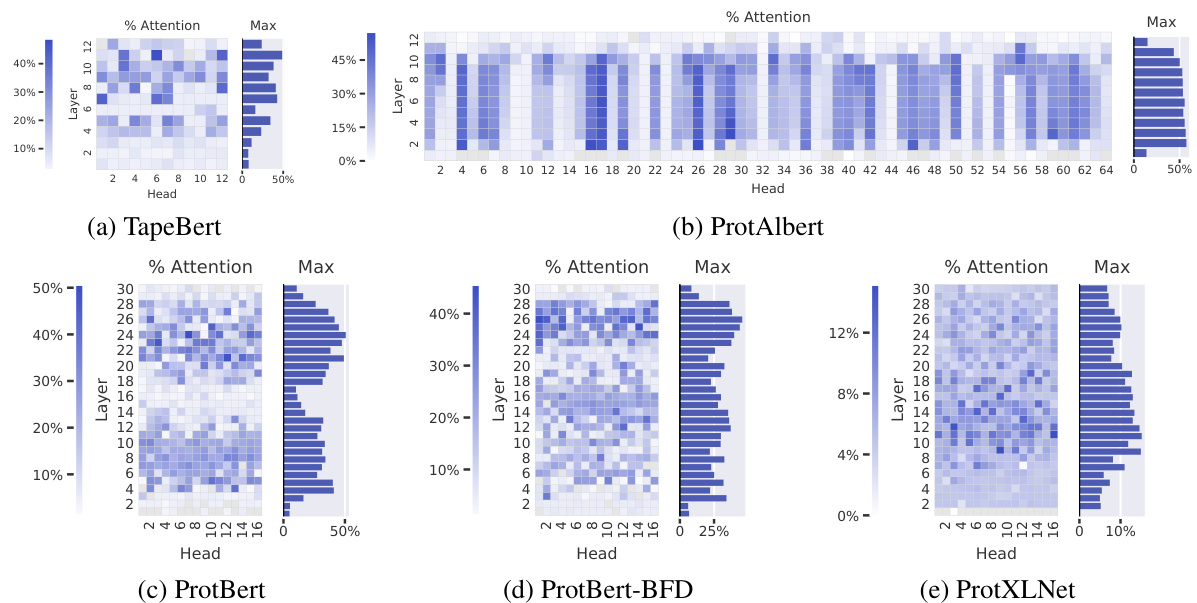
Figure 3: Proportion of attention focused on binding sites across five pretrained models. The heatmaps show the proportion of high-confidence attention $(\alpha_ {i,j}>\theta)$ from each head that is directed to binding sites. In TapeBert (a), for example, we can see that $49%$ of attention in head 11-6 (the 11th layer’s 6th head) is directed to binding sites. The bar plots show the maximum value from each layer.
图 3: 五个预训练模型中对结合位点的注意力分布比例。热力图展示了每个注意力头中指向结合位点的高置信度注意力比例 $(\alpha_ {i,j}>\theta)$ 。以TapeBert (a)为例,可见第11层第6个头(head 11-6)中49%的注意力指向结合位点。条形图展示了每层的最大值。
Considering the models were trained on language modeling tasks without any spatial information, the presence of these structurally-aware attention heads is intriguing. One possible reason for this emergent behavior is that contacts are more likely to bio chemically interact with one another, creating statistical dependencies between the amino acids in contact. By focusing attention on the contacts of a masked position, the language models may acquire valuable context for token prediction.
考虑到这些模型是在没有任何空间信息的语言建模任务上训练的,这些具有结构感知能力的注意力头的存在令人着迷。这种行为涌现的一个可能原因是,接触的氨基酸更有可能发生生化相互作用,从而在接触的氨基酸之间产生统计依赖性。通过将注意力集中在被遮蔽位置的接触上,语言模型可能获得对token预测有价值的上下文信息。
While there seems to be a strong correlation between the attention head output and classically-defined contacts, there are also differences. The models may have learned differing contextual i zed or nuanced formulations that describe amino acid interactions. These learned interactions could then be used for further discovery and investigation or repurposed for prediction tasks similar to how principles of coevolution enabled a powerful representation for structure prediction.
虽然注意力头输出与传统定义的接触之间似乎存在很强的相关性,但也存在差异。这些模型可能学会了描述氨基酸相互作用的不同情境化或细微差别的表达方式。这些习得的相互作用可以用于进一步的发现和研究,或者重新用于类似于共进化原理为结构预测提供强大表征的预测任务。
4.2 BINDING SITES AND POST-TRANSLATIONAL MODIFICATIONS
4.2 结合位点与翻译后修饰
We also analyze how attention interacts with binding sites and post-translational modifications (PTMs), which both play a key role in protein function.
我们还分析了注意力机制如何与结合位点及翻译后修饰 (PTM) 相互作用,这两者在蛋白质功能中都起着关键作用。
Attention targets binding sites throughout most layers of the models. Figure 3 shows the proportion of attention focused on binding sites (Eq. 1) across the heads of the 5 models studied. Attention to binding sites is most pronounced in the ProtAlbert model (Figure 3b), which has 22 heads that focus over $50%$ of attention on bindings sites, whereas the background frequency of binding sites in the dataset is $4.8%$ . The three BERT models (Figures 3a, 3c, and 3d) also attend strongly to binding sites, with attention heads focusing up to $48.2%$ , $50.7%$ , and $45.6%$ of attention on binding sites, respectively. Figure 1b visualizes the attention in one strongly-aligned head from the TapeBert model. Statistical significance tests and a comparison to a null model are provided in Appendix C.3.
注意力机制在模型的大多数层中针对结合位点。图3展示了研究的5个模型各注意力头聚焦于结合位点的比例(式1)。ProtAlbert模型(图3b)对结合位点的注意力最为显著,其22个注意力头将超过$50%$的注意力集中在结合位点上,而数据集中结合位点的背景频率仅为$4.8%$。三个BERT模型(图3a、3c和3d)同样对结合位点表现出强烈关注,其注意力头分别将$48.2%$、$50.7%$和$45.6%$的注意力集中于结合位点。图1b可视化展示了TapeBert模型中一个强对齐注意力头的注意力分布。统计显著性检验及与零模型的对比详见附录C.3。
ProtXLNet (Figure 3e) also targets binding sites, but not as strongly as the other models: the most aligned head focuses $15.1%$ of attention on binding sites, and the average head directs just $6.2%$ of attention to binding sites, compared to $13.2%$ , $19.8%$ , $16.0%$ , and $15.1%$ for the first four models in Figure 3. It’s unclear whether this disparity is due to differences in architectures or pre-training objectives; for example, ProtXLNet uses a bidirectional auto-regressive pre training method (see Appendix A.2), whereas the other 4 models all use masked language modeling objectives.
ProtXLNet (图 3e) 同样以结合位点为靶点,但效果不如其他模型显著:对齐性最强的注意力头仅将 15.1% 的注意力分配给结合位点,而平均注意力头对结合位点的关注度仅为 6.2%。相比之下,图 3 中前四个模型的对应数据分别为 13.2%、19.8%、16.0% 和 15.1%。这种差异是否源于架构差异或预训练目标尚不明确:例如 ProtXLNet 采用双向自回归预训练方法 (参见附录 A.2),而其他四个模型均使用掩码语言建模目标。
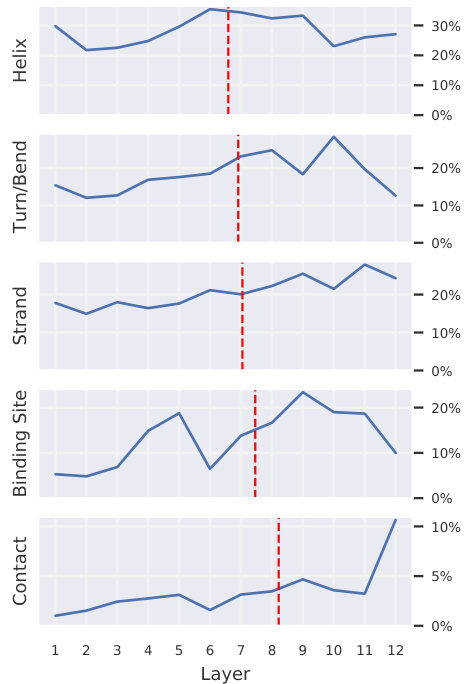
Figure 4: Each plot shows the percentage of attention focused on the given property, averaged over all heads within each layer. The plots, sorted by center of gravity (red dashed line), show that heads in deeper layers focus relatively more attention on binding sites and contacts, whereas attention toward specific secondary structures is more even across layers.
图 4: 每张图显示了各层内所有注意力头对给定属性的平均关注百分比。按重心(红色虚线)排序的图表表明,深层注意力头相对更关注结合位点和接触点,而对特定二级结构的注意力在各层间分布更为均匀。

Figure 5: Performance of probing class if i ers by layer, sorted by task order in Figure 4. The embedding probes (orange) quantify the knowledge of the given property that is encoded in each layer’s output embeddings. The attention probe (blue), show the amount of information encoded in attention weights for the (pairwise) contact feature. Additional details are provided in Appendix B.3.
图 5: 按层探测分类器的性能,按图4中的任务顺序排序。嵌入探针(orange)量化了每层输出嵌入中对给定属性的编码知识。注意力探针(blue)显示了注意力权重中编码的(成对)接触特征信息量。更多细节见附录B.3。
Why does attention target binding sites? In contrast to contact maps, which reveal relationships within proteins, binding sites describe how a protein interacts with other molecules. These external interactions ultimately define the high-level function of the protein, and thus binding sites remain conserved even when the sequence as a whole evolves (Kinjo & Nakamura, 2009). Further, structural motifs in binding sites are mainly restricted to specific families or superfamilies of proteins (Kinjo & Nakamura, 2009), and binding sites can reveal evolutionary relationships among proteins (Lee et al., 2017). Thus binding sites may provide the model with a high-level characterization of the protein that is robust to individual sequence variation. By attending to these regions, the model can leverage this higher-level context when predicting masked tokens throughout the sequence.
注意力为何聚焦于结合位点?与揭示蛋白质内部关系的接触图不同,结合位点描述了蛋白质如何与其他分子相互作用。这些外部相互作用最终决定了蛋白质的高级功能,因此即使整个序列发生进化,结合位点仍保持保守 (Kinjo & Nakamura, 2009)。此外,结合位点的结构基序主要局限于特定蛋白质家族或超家族 (Kinjo & Nakamura, 2009),且结合位点能揭示蛋白质间的进化关系 (Lee et al., 2017)。因此,结合位点可能为模型提供对单个序列变异具有鲁棒性的蛋白质高级特征表征。通过关注这些区域,模型在预测整个序列中被遮蔽的token时,可以利用这种更高层次的上下文信息。
Attention targets PTMs in a small number of heads. A small number of heads in each model concentrate their attention very strongly on amino acids associated with post-translational modifications (PTMs). For example, Head 11-6 in TapeBert focused $64%$ of attention on PTM positions, though these occur at only $0.8%$ of sequence positions in the dataset.3 Similar to our discussion on binding sites, PTMs are critical to protein function (Rubin & Rosen, 1975) and thereby are likely to exhibit behavior that is conserved across the sequence space. See Appendix C.4 for full results.
注意力机制在少数头部中聚焦于翻译后修饰(PTM)位点。每个模型中的少量注意力头会高度集中于与翻译后修饰相关的氨基酸。例如,TapeBert模型的11-6号头部将64%的注意力分配给了PTM位点,尽管这些位点在数据集中仅占序列位置的0.8%。与我们对结合位点的讨论类似,PTM对蛋白质功能至关重要 (Rubin & Rosen, 1975),因此可能在序列空间中表现出保守的行为特性。完整结果参见附录C.4。
4.3 CROSS-LAYER ANALYSIS
4.3 跨层分析
We analyze how attention captures properties of varying complexity across different layers of TapeBert, and compare this to a probing analysis of embeddings and attention weights (see Section 3).
我们分析了TapeBert不同层中注意力机制如何捕捉不同复杂度的属性,并与嵌入向量和注意力权重的探测分析进行了对比(参见第3节)。
Attention targets higher-level properties in deeper layers. As shown in Figure 4, deeper layers focus relatively more attention on binding sites and contacts (high-level concept), whereas secondary structure (low- to mid-level concept) is targeted more evenly across layers. The probing analysis of attention (Figure 5, blue) similarly shows that knowledge of contact maps (a pairwise feature)
注意力在深层针对更高层次的属性。如图4所示,深层相对更关注结合位点和接触点(高层次概念),而二级结构(中低层次概念)在各层的关注分布更为均匀。对注意力的探测分析(图5,蓝色部分)同样显示,接触图(成对特征)的知识...
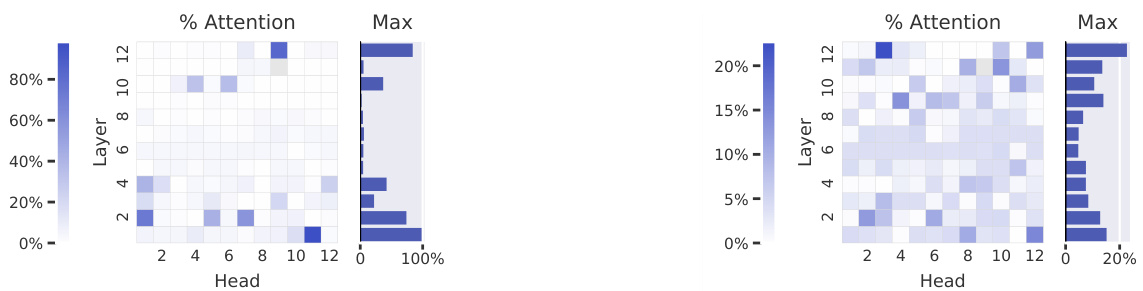
Figure 6: Percentage of each head’s attention focused on amino acids Pro (left) and Phe (right).
图 6: 各注意力头聚焦于氨基酸 Pro (左) 和 Phe (右) 的百分比分布。

Figure 7: Pairwise attention similarity (left) vs. substitution matrix (right) (codes in App. C.5)
图 7: 配对注意力相似度 (左) vs. 替换矩阵 (右) (代码见附录 C.5)
is encoded in attention weights primarily in the last 1-2 layers. These results are consistent with prior work in NLP that suggests deeper layers in text-based Transformers attend to more complex properties (Vig & Belinkov, 2019) and encode higher-level representations (Raganato & Tiedemann, 2018; Peters et al., 2018; Tenney et al., 2019; Jawahar et al., 2019).
主要编码在最后1-2层的注意力权重中。这些结果与NLP领域先前的研究一致,表明基于文本的Transformer更深层关注更复杂的特性 (Vig & Belinkov, 2019) 并编码更高层次的表征 (Raganato & Tiedemann, 2018; Peters et al., 2018; Tenney et al., 2019; Jawahar et al., 2019)。
The embedding probes (Figure 5, orange) also show that the model first builds representations of local secondary structure in lower layers before fully encoding binding sites and contact maps in deeper layers. However, this analysis also reveals stark differences in how knowledge of contact maps is accrued in embeddings, which accumulate this knowledge gradually over many layers, compared to attention weights, which acquire this knowledge only in the final layers in this case. This example points out limitations of common layerwise probing approaches that only consider embeddings, which, intuitively, represent what the model knows but not necessarily how it operational ize s that knowledge.
嵌入探针 (图 5, 橙色) 也显示模型先在较低层构建局部二级结构的表征,随后才在更深层完全编码结合位点和接触图。然而该分析还揭示了接触图知识在嵌入向量与注意力权重中的积累方式存在显著差异:嵌入向量通过多个层级逐步积累这类知识,而本例中注意力权重仅在最后几层才获取该知识。这个例子指出了常见层级探测方法的局限性——它们仅考虑嵌入向量 (直观上反映模型知道什么) 而忽略了模型如何运用这些知识。
4.4 AMINO ACIDS AND THE SUBSTITUTION MATRIX
4.4 氨基酸与替换矩阵
In addition to high-level structural and functional properties, we also performed a fine-grained analysis of the interaction between attention and particular amino acids.
除了高层次的结构和功能特性外,我们还对注意力机制与特定氨基酸之间的相互作用进行了细粒度分析。
Attention heads specialize in particular amino acids. We computed the proportion of TapeBert’s attention to each of the 20 standard amino acids, as shown in Figure 6 for two example amino acids. For 16 of the amino acids, there exists an attention head that focuses over $25%$ of attention on that amino acid, significantly greater than the background frequencies of the corresponding amino acids, which range from $1.3%$ to $9.4%$ . Similar behavior was observed for ProtBert, ProtBert-BFD, ProtAlbert, and ProtXLNet models, with 17, 15, 16, and 18 amino acids, respectively, receiving greater than $25%$ of the attention from at least one attention head. Detailed results for TapeBert including statistical significance tests and comparison to a null model are presented in Appendix C.5.
注意力头专注于特定氨基酸。我们计算了TapeBert对20种标准氨基酸中每一种的关注比例,图6展示了两例氨基酸的情况。在16种氨基酸中,存在至少一个注意力头对其关注度超过25%,显著高于这些氨基酸1.3%至9.4%的背景频率。ProtBert、ProtBert-BFD、ProtAlbert和ProtXLNet模型也表现出类似行为,分别有17、15、16和18种氨基酸获得至少一个注意力头超过25%的关注度。TapeBert的详细结果(包括统计显著性检验及与零模型的对比)见附录C.5。
Attention is consistent with substitution relationships. A natural follow-up question from the above analysis is whether each head has “memorized” specific amino acids to target, or whether it has actually learned meaningful properties that correlate with particular amino acids. To test the latter hypothesis, we analyze whether amino acids with similar structural and functional properties are attended to similarly across heads. Specifically, we compute the Pearson correlation between the distribution of attention across heads between all pairs of distinct amino acids, as shown in Figure 7 (left) for TapeBert. For example, the entry for Pro (P) and Phe (F) is the correlation between the two heatmaps in Figure 6. We compare these scores to the BLOSUM62 substitution scores (Sec. 2) in Figure 7 (right), and find a Pearson correlation of 0.73, suggesting that attention is moderately consistent with substitution relationships. Similar correlations are observed for the ProtTrans models: 0.68 (ProtBert), 0.75 (ProtBert-BFD), 0.60 (ProtAlbert), and 0.71 (ProtXLNet). As a baseline, the randomized versions of these models (Appendix B.2) yielded correlations of -0.02 (TapeBert), 0.02 (ProtBert), -0.03 (ProtBert-BFD), -0.05 (ProtAlbert), and 0.21 (ProtXLNet).
注意力机制与替换关系具有一致性。基于上述分析,一个自然的后续问题是:每个注意力头是否"记忆"了特定的目标氨基酸,还是真正学习了与特定氨基酸相关的有意义属性。为验证后一种假设,我们分析具有相似结构和功能特性的氨基酸在不同注意力头中是否获得相似的关注。具体而言,我们计算所有不同氨基酸对在注意力头间的注意力分布皮尔逊相关性,如TapeBert的图7(左)所示。例如,Pro(P)与Phe(F)的对应值即为图6中两个热力图的相关性。我们将这些分数与BLOSUM62替换分数(第2节)在图7(右)中进行比较,发现皮尔逊相关系数为0.73,表明注意力机制与替换关系存在适度一致性。ProtTrans模型也观察到类似相关性:0.68(ProtBert)、0.75(ProtBert-BFD)、0.60(ProtAlbert)和0.71(ProtXLNet)。作为基线,这些模型的随机化版本(附录B.2)产生的相关性分别为-0.02(TapeBert)、0.02(ProtBert)、-0.03(ProtBert-BFD)、-0.05(ProtAlbert)和0.21(ProtXLNet)。
5 RELATED WORK
5 相关工作
5.1 PROTEIN LANGUAGE MODELS
5.1 蛋白质语言模型
Deep neural networks for protein language modeling have received broad interest. Early work applied the Skip-gram model (Mikolov et al., 2013) to construct continuous embeddings from protein sequences (Asgari & Mofrad, 2015). Sequence-only language models have since been trained through auto regressive or auto encoding self-supervision objectives for disc rim i native and generative tasks, for example, using LSTMs or Transformer-based architectures (Alley et al., 2019; Bepler & Berger, 2019; Rao et al., 2019; Rives et al., 2019). TAPE created a benchmark of five tasks to assess protein sequence models, and ProtTrans also released several large-scale pretrained protein Transformer models (Elnaggar et al., 2020). Riesselman et al. (2019); Madani et al. (2020) trained auto regressive generative models to predict the functional effect of mutations and generate natural-like proteins.
用于蛋白质语言建模的深度神经网络已引起广泛关注。早期研究应用Skip-gram模型(Mikolov等人,2013)从蛋白质序列构建连续嵌入表示(Asgari & Mofrad,2015)。此后,仅基于序列的语言模型通过自回归或自编码的自监督目标进行训练,用于判别性和生成性任务,例如使用LSTM或基于Transformer的架构(Alley等人,2019;Bepler & Berger,2019;Rao等人,2019;Rives等人,2019)。TAPE建立了包含五项任务的基准测试来评估蛋白质序列模型,ProtTrans也发布了多个大规模预训练的蛋白质Transformer模型(Elnaggar等人,2020)。Riesselman等人(2019)和Madani等人(2020)训练了自回归生成模型来预测突变的功能效应并生成类天然蛋白质。
From an interpret ability perspective, Rives et al. (2019) showed that the output embeddings from a pretrained Transformer can recapitulate structural and functional properties of proteins through learned linear transformations. Various works have analyzed output embeddings of protein models through dimensionality reduction techniques such as PCA or t-SNE (Elnaggar et al., 2020; Biswas et al., 2020). In our work, we take an interpret ability-first perspective to focus on the internal model representations, specifically attention and intermediate hidden states, across multiple protein language models. We also explore novel biological properties including binding sites and post-translational modifications.
从可解释性角度来看,Rives等人(2019)研究表明,预训练Transformer的输出嵌入(embedding)可通过学习到的线性变换重现蛋白质的结构与功能特性。多项工作通过PCA或t-SNE等降维技术分析了蛋白质模型的输出嵌入(Elnaggar等人,2020;Biswas等人,2020)。在本研究中,我们采用"可解释性优先"的视角,重点关注多个蛋白质语言模型的内部表征(representation),特别是注意力机制(attention)和中间隐藏状态(hidden states)。我们还探索了包括结合位点(binding sites)和翻译后修饰(post-translational modifications)在内的新型生物学特性。
5.2 INTERPRETING MODELS IN NLP
5.2 NLP 中的模型解释
The rise of deep neural networks in ML has also led to much work on interpreting these so-called black-box models. This section reviews the NLP interpret ability literature on the Transformer model, which is directly comparable to our work on interpreting Transformer models of protein sequences.
深度学习神经网络在机器学习领域的兴起也催生了许多关于解释这些所谓"黑盒模型"的研究。本节回顾了针对Transformer模型的自然语言处理可解释性文献,这些研究与我们在蛋白质序列Transformer模型解释方面的工作具有直接可比性。
Interpreting Transformers. The Transformer is a neural architecture that uses attention to accelerate learning (Vaswani et al., 2017). In NLP, transformers are the backbone of state-of-the-art pre-trained language models such as BERT (Devlin et al., 2019). BERTology focuses on interpreting what the BERT model learns about language using a suite of probes and interventions (Rogers et al., 2020). So-called diagnostic class if i ers are used to interpret the outputs from BERT’s layers (Veldhoen et al., 2016). At a high level, mechanisms for interpreting BERT can be placed into three main categories: interpreting the learned embeddings (Ethayarajh, 2019; Wiedemann et al., 2019; Mickus et al., 2020; Adi et al., 2016; Conneau et al., 2018), BERT’s learned knowledge of syntax (Lin et al., 2019; Liu et al., 2019; Tenney et al., 2019; Htut et al., 2019; Hewitt & Manning, 2019; Goldberg, 2019), and BERT’s learned knowledge of semantics (Tenney et al., 2019; Ettinger, 2020).
解读Transformer。Transformer是一种利用注意力机制加速学习的神经网络架构 (Vaswani et al., 2017)。在自然语言处理领域,Transformer已成为BERT (Devlin et al., 2019) 等最先进预训练语言模型的核心组件。BERTology研究通过一系列探测和干预手段来解析BERT模型对语言的学习机制 (Rogers et al., 2020),其中诊断分类器被用于解释BERT各层的输出 (Veldhoen et al., 2016)。总体而言,BERT的解读机制可分为三大类:解析学习到的嵌入表示 (Ethayarajh, 2019; Wiedemann et al., 2019; Mickus et al., 2020; Adi et al., 2016; Conneau et al., 2018)、模型习得的句法知识 (Lin et al., 2019; Liu et al., 2019; Tenney et al., 2019; Htut et al., 2019; Hewitt & Manning, 2019; Goldberg, 2019),以及模型掌握的语义知识 (Tenney et al., 2019; Ettinger, 2020)。
Interpreting attention specifically. Interpreting attention on textual sequences is a wellestablished area of research (Wiegreffe & Pinter, 2019; Zhong et al., 2019; Brunner et al., 2020; Hewitt & Manning, 2019). Past work has been shown that attention correlates with syntactic and semantic relationships in natural language in some cases (Clark et al., 2019; Vig & Belinkov, 2019; Htut et al., 2019). Depending on the task and model architecture, attention may have less or more explanatory power for model predictions (Jain & Wallace, 2019; Serrano & Smith, 2019; Pruthi et al., 2020; Moradi et al., 2019; Vashishth et al., 2019). Visualization techniques have been used to convey the structure and properties of attention in Transformers (Vaswani et al., 2017; Kovaleva et al., 2019; Hoover et al., 2020; Vig, 2019). Recent work has begun to analyze attention in Transformer models outside of the domain of natural language (Schwaller et al., 2020; Payne et al., 2020).
解读注意力机制。针对文本序列的注意力机制解读已形成成熟的研究领域 (Wiegreffe & Pinter, 2019; Zhong et al., 2019; Brunner et al., 2020; Hewitt & Manning, 2019)。研究表明,注意力机制在某些情况下与自然语言的句法和语义关系存在关联 (Clark et al., 2019; Vig & Belinkov, 2019; Htut et al., 2019)。根据任务和模型架构的差异,注意力机制对模型预测的解释力可能强弱不一 (Jain & Wallace, 2019; Serrano & Smith, 2019; Pruthi et al., 2020; Moradi et al., 2019; Vashishth et al., 2019)。可视化技术已被广泛应用于展现Transformer中注意力机制的结构与特性 (Vaswani et al., 2017; Kovaleva et al., 2019; Hoover et al., 2020; Vig, 2019)。最新研究开始探索自然语言领域之外Transformer模型的注意力机制分析 (Schwaller et al., 2020; Payne et al., 2020)。
Our work extends these methods to protein sequence models by considering particular bio physical properties and relationships. We also present a joint cross-layer probing analysis of attention weights and layer embeddings. While past work in NLP has analyzed attention and embeddings across layers, we believe we are the first to do so in any domain using a single, unified metric, which enables us to directly compare the relative information content of the two representations. Finally, we present a novel tool for visualizing attention embedded in three-dimensional structure.
我们的工作通过考虑特定的生物物理特性和关系,将这些方法扩展到蛋白质序列模型。我们还提出了一种对注意力权重和层嵌入进行联合跨层探测分析的方法。虽然过去在自然语言处理(NLP)领域已有研究分析了跨层的注意力和嵌入,但我们相信我们是首个在任何领域使用单一统一指标进行此类分析的研究,这使我们能够直接比较两种表征的相对信息量。最后,我们提出了一种新颖的可视化工具,用于展示三维结构中嵌入的注意力。
6 CONCLUSIONS AND FUTURE WORK
6 结论与未来工作
This paper builds on the synergy between NLP and computational biology by adapting and extending NLP interpret ability methods to protein sequence modeling. We show how a Transformer language model recovers structural and functional properties of proteins and integrates this knowledge directly into its attention mechanism. While this paper focuses on reconciling attention with known properties of proteins, one might also leverage attention to uncover novel relationships or more nuanced forms of existing measures such as contact maps, as discussed in Section 4.1. In this way, language models have the potential to serve as tools for scientific discovery. But in order for learned representations to be accessible to domain experts, they must be presented in an appropriate context to facilitate discovery. Visualizing attention in the context of protein structure (Figure 1) is one attempt to do so. We believe there is the potential to develop such contextual visualization s of learned representations in a range of scientific domains.
本文基于自然语言处理(NLP)与计算生物学的协同效应,将NLP可解释性方法适配并扩展到蛋白质序列建模领域。我们展示了Transformer语言模型如何重构蛋白质的结构与功能特性,并将这些知识直接整合到其注意力机制中。虽然本文主要关注将注意力机制与已知蛋白质特性相印证,但如第4.1节所述,人们也可以利用注意力机制来揭示新的关联,或对现有测量指标(如接触图)进行更精细的刻画。通过这种方式,语言模型有望成为科学发现的工具。但为了让领域专家能够理解学习到的表征,必须将其置于合适的上下文中以促进发现。在蛋白质结构背景下可视化注意力机制(图1)就是一次这样的尝试。我们相信在一系列科学领域中,这种针对学习表征的上下文可视化方法都具有发展潜力。
ACKNOWLEDGMENTS
致谢
We would like to thank Xi Victoria Lin, Stephan Zheng, Melvin Gruesbeck, and the anonymous reviewers for their valuable feedback.
感谢Xi Victoria Lin、Stephan Zheng、Melvin Gruesbeck以及匿名评审人员提供的宝贵意见。
REFERENCES
参考文献
Alexis Conneau, German Kruszewski, Guillaume Lample, Loïc Barrault, and Marco Baroni. What you can cram into a single $\$1\$ &!#* vector: Probing sentence embeddings for linguistic properties. In Proceedings of the 56th Annual Meeting of the Association for Computational Linguistics, pp. 2126–2136, 2018.
Alexis Conneau、German Kruszewski、Guillaume Lample、Loïc Barrault 和 Marco Baroni。你能塞进单个 $\$1\$ &!#* 向量的东西:探究句子嵌入的语言特性。载于《第56届计算语言学协会年会论文集》,第2126–2136页,2018年。
Jacob Devlin, Ming-Wei Chang, Kenton Lee, and Kristina Toutanova. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers), pp. 4171–4186, Minneapolis, Minnesota, 2019. Association for Computational Linguistics.
Jacob Devlin、Ming-Wei Chang、Kenton Lee 和 Kristina Toutanova。BERT: 面向语言理解的深度双向Transformer预训练。载于《2019年北美计算语言学协会人类语言技术会议论文集》(长篇与短篇论文),第1卷,第4171–4186页,明尼苏达州明尼阿波利斯市,2019年。计算语言学协会。
Sara El-Gebali, Jaina Mistry, Alex Bateman, Sean R. Eddy, Aurélien Luciani, Simon C. Potter, Matloob Qureshi, Lorna J. Richardson, Gustavo A. Salazar, Alfredo Smart, Erik L. L. Sonnhammer, Layla Hirsh, Lisanna Paladin, Damiano Piovesan, Silvio C. E. Tosatto, and Robert D. Finn. The Pfam protein families database in 2019. Nucleic Acids Research, 47(D1):D427–D432, January 2019a. doi: 10.1093/nar/gky995.
Sara El-Gebali、Jaina Mistry、Alex Bateman、Sean R. Eddy、Aurélien Luciani、Simon C. Potter、Matloob Qureshi、Lorna J. Richardson、Gustavo A. Salazar、Alfredo Smart、Erik L. L. Sonnhammer、Layla Hirsh、Lisanna Paladin、Damiano Piovesan、Silvio C. E. Tosatto 和 Robert D. Finn。2019 年 Pfam 蛋白质家族数据库。核酸研究,47(D1):D427–D432,2019 年 1 月。doi: 10.1093/nar/gky995。
Sara El-Gebali, Jaina Mistry, Alex Bateman, Sean R Eddy, Aurélien Luciani, Simon C Potter, Matloob Qureshi, Lorna J Richardson, Gustavo A Salazar, Alfredo Smart, Erik L L Sonnhammer, Layla Hirsh, Lisanna Paladin, Damiano Piovesan, Silvio C E Tosatto, and Robert D Finn. The Pfam protein families database in 2019. Nucleic Acids Research, 47(D1):D427–D432, 2019b. ISSN 0305-1048. doi: 10.1093/nar/gky995.
Sara El-Gebali、Jaina Mistry、Alex Bateman、Sean R Eddy、Aurélien Luciani、Simon C Potter、Matloob Qureshi、Lorna J Richardson、Gustavo A Salazar、Alfredo Smart、Erik L L Sonnhammer、Layla Hirsh、Lisanna Paladin、Damiano Piovesan、Silvio C E Tosatto 和 Robert D Finn。《2019年Pfam蛋白质家族数据库》。核酸研究(Nucleic Acids Research),47(D1):D427–D432,2019b。ISSN 0305-1048。doi: 10.1093/nar/gky995。
Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rihawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steinegger, Debsindhu Bhowmik, and Burkhard Rost. ProtTrans: Towards cracking the language of life’s code through self-supervised deep learning and high performance computing. arXiv preprint arXiv:2007.06225, 2020.
Ahmed Elnaggar、Michael Heinzinger、Christian Dallago、Ghalia Rihawi、Yu Wang、Llion Jones、Tom Gibbs、Tamas Feher、Christoph Angerer、Martin Steinegger、Debsindhu Bhowmik 和 Burkhard Rost。ProtTrans: 通过自监督深度学习和高性能计算破解生命密码的语言。arXiv预印本 arXiv:2007.06225, 2020。
Kawin Ethayarajh. How contextual are contextual i zed word representations? Comparing the geometry of BERT, ELMo, and GPT-2 embeddings. In Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP), pp. 55–65, Hong Kong, China, 2019. Association for Computational Linguistics.
Kawin Ethayarajh. 语境化词表征的上下文关联性如何?比较BERT、ELMo和GPT-2嵌入的几何特性。载于《2019年自然语言处理经验方法会议暨第九届自然语言处理国际联合会议论文集》(EMNLP-IJCNLP),第55-65页,中国香港,2019年。计算语言学协会。
Allyson Ettinger. What BERT is not: Lessons from a new suite of psycho linguistic diagnostics for language models. Transactions of the Association for Computational Linguistics, 8:34–48, 2020.
Allyson Ettinger. 什么是BERT所不具备的:从一套新的心理语言学诊断工具看大语言模型的启示。Transactions of the Association for Computational Linguistics, 8:34–48, 2020.
Naomi K Fox, Steven E Brenner, and John-Marc Chandonia. SCOPe: Structural classification of proteins—extended, integrating scop and astral data and classification of new structures. Nucleic Acids Research, 42(D1):D304–D309, 2013.
Naomi K Fox、Steven E Brenner 和 John-Marc Chandonia。SCOPe: 蛋白质结构分类扩展版——整合 SCOP 与 ASTRAL 数据及新结构分类。《核酸研究》,42(D1):D304–D309,2013年。
Yoav Goldberg. Assessing BERT’s syntactic abilities. arXiv preprint arXiv:1901.05287, 2019.
Yoav Goldberg. Assessing BERT’s syntactic abilities. arXiv preprint arXiv:1901.05287, 2019.
Christopher Grimsley, Elijah Mayfield, and Julia R.S. Bursten. Why attention is not explanation: Surgical intervention and causal reasoning about neural models. In Proceedings of The 12th Language Resources and Evaluation Conference, pp. 1780–1790, Marseille, France, May 2020. European Language Resources Association. ISBN 979-10-95546-34-4. URL https://www. aclweb.org/anthology/2020.lrec-1.220.
Christopher Grimsley、Elijah Mayfield 和 Julia R.S. Bursten。为什么注意力不是解释:神经模型的手术干预与因果推理。载于《第十二届语言资源与评估会议论文集》,第1780-1790页,法国马赛,2020年5月。欧洲语言资源协会。ISBN 979-10-95546-34-4。URL https://www.aclweb.org/anthology/2020.lrec-1.220。
S Henikoff and J G Henikoff. Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences, 89(22):10915–10919, 1992. ISSN 0027-8424. doi: 10.1073/ pnas.89.22.10915. URL https://www.pnas.org/content/89/22/10915.
S Henikoff 和 J G Henikoff. 基于蛋白质块的氨基酸替换矩阵. Proceedings of the National Academy of Sciences, 89(22):10915–10919, 1992. ISSN 0027-8424. doi: 10.1073/pnas.89.22.10915. URL https://www.pnas.org/content/89/22/10915.
John Hewitt and Christopher D Manning. A structural probe for finding syntax in word representations. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers), pp. 4129–4138, 2019.
John Hewitt 和 Christopher D Manning. 一种用于在词表示中寻找句法结构的结构探针. 载于《2019年北美计算语言学协会人类语言技术会议论文集》第1卷(长文与短文), 第4129–4138页, 2019年.
Benjamin Hoover, Hendrik Strobelt, and Sebastian Gehrmann. exBERT: A Visual Analysis Tool to Explore Learned Representations in Transformer Models. In Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics: System Demonstrations, pp. 187–196. Association for Computational Linguistics, 2020.
Benjamin Hoover、Hendrik Strobelt 和 Sebastian Gehrmann. exBERT: 一种用于探索Transformer模型学习表征的可视化分析工具. 载于《第58届计算语言学协会年会系统演示论文集》第187-196页. 计算语言学协会, 2020.
Phu Mon Htut, Jason Phang, Shikha Bordia, and Samuel R Bowman. Do attention heads in BERT track syntactic dependencies? arXiv preprint arXiv:1911.12246, 2019.
Phu Mon Htut、Jason Phang、Shikha Bordia 和 Samuel R Bowman。BERT 中的注意力头是否追踪句法依赖关系?arXiv 预印本 arXiv:1911.12246,2019。
Pooya Moradi, Nishant Kambhatla, and Anoop Sarkar. Interrogating the explanatory power of attention in neural machine translation. In Proceedings of the 3rd Workshop on Neural Generation and Translation, pp. 221–230, Hong Kong, November 2019. Association for Computational Linguistics. doi: 10.18653/v1/D19-5624. URL https://www.aclweb.org/anthology/ D19-5624.
Pooya Moradi、Nishant Kambhatla 和 Anoop Sarkar。探究注意力机制在神经机器翻译中的解释力。载于《第三届神经生成与翻译研讨会论文集》,第221-230页,香港,2019年11月。计算语言学协会。doi: 10.18653/v1/D19-5624。URL https://www.aclweb.org/anthology/D19-5624。
Sarah Wiegreffe and Yuval Pinter. Attention is not not explanation. In Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP), pp. 11–20, November 2019.
Sarah Wiegreffe 和 Yuval Pinter. Attention 并非解释。载于《2019年自然语言处理实证方法会议暨第九届自然语言处理国际联合会议论文集》(EMNLP-IJCNLP),第11-20页,2019年11月。
Zhilin Yang, Zihang Dai, Yiming Yang, Jaime Carbonell, Russ R Salak hut dino v, and Quoc V Le. XLNet: Generalized auto regressive pre training for language understanding. In H. Wallach, H. Larochelle, A. Bey gel zi mer, F. d'Alché-Buc, E. Fox, and R. Garnett (eds.), Advances in Neural Information Processing Systems, volume 32, pp. 5753–5763. Curran Associates, Inc., 2019.
Zhilin Yang、Zihang Dai、Yiming Yang、Jaime Carbonell、Russ R Salakhutdinov 和 Quoc V Le。XLNet: 语言理解的广义自回归预训练。见 H. Wallach、H. Larochelle、A. Beygelzimer、F. d'Alché-Buc、E. Fox 和 R. Garnett (编), 《神经信息处理系统进展》, 第 32 卷, 第 5753–5763 页。Curran Associates 公司, 2019。
Ruiqi Zhong, Steven Shao, and Kathleen McKeown. Fine-grained sentiment analysis with faithful attention. arXiv preprint arXiv:1908.06870, 2019.
Ruiqi Zhong, Steven Shao, and Kathleen McKeown. 基于忠实注意力的细粒度情感分析. arXiv preprint arXiv:1908.06870, 2019.
A MODEL OVERVIEW
模型概述
A.1 PRE-TRAINED MODELS
A.1 预训练模型
Table 1 provides an overview of the five pre-trained Transformer models studied in this work. The models originate from the TAPE and ProtTrans repositories, spanning three model architectures: BERT, ALBERT, and XLNet.
表 1: 概述了本研究中涉及的五个预训练Transformer模型。这些模型来源于TAPE和ProtTrans代码库,涵盖三种模型架构:BERT、ALBERT和XLNet。
Table 1: Summary of pre-trained models analyzed, including the source of the model, the type of Transformer used, the number of layers and heads, the total number of model parameters, the source of the pre-training dataset, and the number of protein sequences in the pre-training dataset.
| Source | Name | Type | Layers | Heads | Params | Train Dataset | #Seq |
| TAPE | TapeBert | BERT | 12 | 12 | 94M | Pfam | 31M |
| ProtTrans | ProtBert | BERT | 30 | 16 | 420M | Uniref100 | 216M |
| ProtTrans | ProtBert-BFD | BERT | 30 | 16 | 420M | BFD | 2.1B |
| ProtTrans | ProtAlbert | ALBERT | 12 | 64 | 224M | Uniref100 | 216M |
| ProtTrans | ProtXLNet | XLNet | 30 | 16 | 409M | Uniref100 | 216M |
表 1: 分析的预训练模型汇总,包括模型来源、使用的Transformer类型、层数和头数、模型参数总数、预训练数据集来源以及预训练数据集中的蛋白质序列数量。
| 来源 | 名称 | 类型 | 层数 | 头数 | 参数 | 训练数据集 | 序列数 |
|---|---|---|---|---|---|---|---|
| TAPE | TapeBert | BERT | 12 | 12 | 94M | Pfam | 31M |
| ProtTrans | ProtBert | BERT | 30 | 16 | 420M | Uniref100 | 216M |
| ProtTrans | ProtBert-BFD | BERT | 30 | 16 | 420M | BFD | 2.1B |
| ProtTrans | ProtAlbert | ALBERT | 12 | 64 | 224M | Uniref100 | 216M |
| ProtTrans | ProtXLNet | XLNet | 30 | 16 | 409M | Uniref100 | 216M |
A.2 BERT TRANSFORMER ARCHITECTURE
A.2 BERT Transformer架构
Stacked Encoder: BERT uses a stacked-encoder architecture, which inputs a sequence of tokens ${\pmb x}=(x_ {1},...,x_ {n})$ and applies position and token embeddings followed by a series of encoder layers. Each layer applies multi-head self-attention (see below) in combination with a feed forward network, layer normalization, and residual connections. The output of each layer $\ell$ is a sequence of contextual i zed embeddings $(\mathbf{h}_ {1}^{(\ell)},\dots,\mathbf{h}_ {n}^{(\ell)})$ .
堆叠编码器:BERT采用堆叠编码器架构,其输入为一个Token序列 ${\pmb x}=(x_ {1},...,x_ {n})$ ,先经过位置嵌入和Token嵌入处理,再通过一系列编码器层。每层结合多头自注意力机制(详见下文)、前馈网络、层归一化及残差连接进行处理。每层 $\ell$ 的输出为一组上下文嵌入向量 $(\mathbf{h}_ {1}^{(\ell)},\dots,\mathbf{h}_ {n}^{(\ell)})$ 。
Self-Attention: Given an input $\pmb{x}=(x_ {1},\dots,x_ {n})$ , the self-attention mechanism assigns to each token pair $i,j$ an attention weight $\alpha_ {i,j}>0$ where $\textstyle\sum_ {j}\alpha_ {i,j}=1$ . Attention in BERT is bidirectional. In the multi-layer, multi-head setting, $\alpha$ is specific to a layer and head. The BERT-Base model has 12 layers and 12 heads. Each attention head learns a distinct set of weights, resulting in $12\mathrm{x}12=144$ distinct attention mechanisms in this case.
自注意力机制:给定输入 $\pmb{x}=(x_ {1},\dots,x_ {n})$,自注意力机制会为每个token对 $i,j$ 分配注意力权重 $\alpha_ {i,j}>0$,其中 $\textstyle\sum_ {j}\alpha_ {i,j}=1$。BERT中的注意力是双向的。在多层多头设置中,$\alpha$ 特定于某一层和某一个头。BERT-Base模型具有12层和12个头。每个注意力头学习一组不同的权重,因此在这种情况下会产生 $12\mathrm{x}12=144$ 种不同的注意力机制。
The attention weights $\alpha_ {i,j}$ are computed from the scaled dot-product of the query vector of $i$ and the key vector of $j$ , followed by a softmax operation. The attention weights are then used to produce a weighted sum of value vectors:
注意力权重 $\alpha_ {i,j}$ 由查询向量 $i$ 与键向量 $j$ 的缩放点积计算得出,随后进行 softmax 运算。这些注意力权重随后用于生成值向量的加权和:
$$
{\mathrm{Attention}}(Q,K,V)=\operatorname{softmax}\left({\frac{Q K^{T}}{\sqrt{d_ {k}}}}\right)V
$$
$$
{\mathrm{Attention}}(Q,K,V)=\operatorname{softmax}\left({\frac{Q K^{T}}{\sqrt{d_ {k}}}}\right)V
$$
using query matrix $Q$ , key matrix $K$ , and value matrix $V$ , where $d_ {k}$ is the dimension of $K$ . In a multi-head setting, the queries, keys, and values are linearly projected $h$ times, and the attention operation is performed in parallel for each representation, with the results concatenated.
使用查询矩阵 $Q$ 、键矩阵 $K$ 和值矩阵 $V$ ,其中 $d_ {k}$ 是 $K$ 的维度。在多头设置中,查询、键和值会被线性投影 $h$ 次,并对每个表示并行执行注意力操作,最后将结果拼接起来。
A.3 OTHER TRANSFORMER VARIANTS
A.3 其他Transformer变体
ALBERT: The architecture of ALBERT differs from BERT in two ways: (1) It shares parameters across layers, unlike BERT which learns distinct parameters for every layer and (2) It uses factorized embeddings, which allows the input token embeddings to be of a different (smaller) size than the hidden states. The original version of ALBERT designed for text also employed a sentence-order prediction pre training task, but this was not used on the models studied in this paper.
ALBERT的架构与BERT有两点不同:(1) 它在各层之间共享参数,而BERT的每一层都学习独立的参数;(2) 它采用因子分解嵌入(factorized embeddings),使得输入token嵌入的维度可以小于隐藏状态的维度。原始文本版ALBERT还使用了句子顺序预测预训练任务,但本文研究的模型未采用该技术。
XLNet: Instead of the masked-language modeling pre training objective use for BERT, XLNet uses a bidirectional auto-regressive pre training method that considers all possible orderings of the input factorization. The architecture also adds a segment recurrence mechanism to process long sequences, as well as a relative rather than absolute encoding scheme.
XLNet:不同于BERT使用的掩码语言建模预训练目标,XLNet采用了一种双向自回归预训练方法,该方法考虑了输入因子分解的所有可能排列顺序。该架构还增加了处理长序列的分段循环机制,以及相对而非绝对的编码方案。
B ADDITIONAL EXPERIMENTAL DETAILS
B 补充实验细节
B.1 ALTERNATIVE ATTENTION AGREEMENT METRIC
B.1 替代注意力一致性指标
Here we present an alternative formulation to Eq. 1 based on an attention-weighted average. We define an indicator function $f(i,j)$ for property $f$ that returns 1 if the property is present in token pair $(i,j)$ (i.e., if amino acids $i$ and $j$ are in contact), and zero otherwise. We then compute the proportion of attention that matches with $f$ over a dataset $\boldsymbol{X}$ as follows:
我们提出一种基于注意力加权平均的公式替代方案。定义属性 $f$ 的指示函数 $f(i,j)$ ,若该属性存在于token对 $(i,j)$ 中(即氨基酸 $i$ 与 $j$ 接触)则返回1,否则返回0。随后计算与属性 $f$ 匹配的注意力在数据集 $\boldsymbol{X}$ 中的占比:
$$
p_ {\alpha}(f)=\sum_ {x\in X}\sum_ {i=1}^{|x|}\sum_ {j=1}^{|x|}f(i,j)\alpha_ {i,j}(x){\bigg/}\sum_ {x\in X}\sum_ {i=1}^{|x|}\sum_ {j=1}^{|x|}\alpha_ {i,j}(x)
$$
$$
p_ {\alpha}(f)=\sum_ {x\in X}\sum_ {i=1}^{|x|}\sum_ {j=1}^{|x|}f(i,j)\alpha_ {i,j}(x){\bigg/}\sum_ {x\in X}\sum_ {i=1}^{|x|}\sum_ {j=1}^{|x|}\alpha_ {i,j}(x)
$$
where $\alpha_ {i,j}(x)$ denotes the attention from $i$ to $j$ for input sequence $x$ .
其中 $\alpha_ {i,j}(x)$ 表示输入序列 $x$ 中从 $i$ 到 $j$ 的注意力权重。
B.2 STATISTICAL SIGNIFICANCE TESTING AND NULL MODELS
B.2 统计显著性检验与零模型
We perform statistical significance tests to determine whether any results based on the metric defined in Equation 1 are due to chance. Given a property $f$ , as defined in Section 3, we perform a twoproportion z-test comparing (1) the proportion of high-confidence attention arcs $(\alpha_ {i,j}>\theta)$ for which $f(i,j)=1$ , and (2) the proportion of all possible pairs $i,j$ for which $f(i,j)=1$ . Note that the first proportion is exactly the metric $p_ {\alpha}(f)$ defined in Equation 1 (e.g. the proportion of attention aligned with contact maps). The second proportion is simply the background frequency of the property (e.g. the background frequency of contacts). Since we extract the maximum scores over all of the heads in the model, we treat this as a case of multiple hypothesis testing and apply the Bonferroni correction, with the number of hypotheses $m$ equal to the number of attention heads.
我们进行统计显著性检验,以确定基于公式1定义的指标得出的任何结果是否具有偶然性。给定第3节中定义的属性$f$,我们执行双比例z检验,比较:(1) 高置信度注意力弧$(\alpha_ {i,j}>\theta)$中满足$f(i,j)=1$的比例,以及(2) 所有可能配对$i,j$中满足$f(i,j)=1$的比例。需要注意的是,第一个比例正是公式1中定义的指标$p_ {\alpha}(f)$(例如与接触图谱对齐的注意力比例)。第二个比例则是该属性的背景频率(例如接触的背景频率)。由于我们从模型所有注意力头中提取最大分数,因此将其视为多重假设检验情况,并应用Bonferroni校正,假设数量$m$等于注意力头的总数。
As an additional check that the results did not occur by chance, we also report results on baseline (null) models. We initially considered using two forms of null models: (1) a model with randomly initialized weights. and (2) a model trained on randomly shuffled sequences. However, in both cases, none of the sequences in the dataset yielded attention weights greater than the attention threshold $\theta$ . This suggests that the mere existence of the high-confidence attention weights used in the analysis could not have occurred by chance, but it does not shed light on the particular analyses performed. Therefore, we implemented an alternative random iz ation scheme in which we randomly shuffle attention weights from the original models as a post-processing step. Specifically, we permute the sequence of attention weights from each token for every attention head. To illustrate, let’s say that the original model produced attention weights of (0.3, 0.2, 0.1, 0.4, 0.0) from position $i$ in protein sequence $x$ from head $h$ , where $|x|=5$ . In the null model, the attention weights from position $i$ in sequence $x$ in head $h$ would be a random permutation of those weights, e.g., (0.2, 0.0, 0.4, 0.3, 0.1). Note that these are still valid attention weights as they would sum to 1 (since the original weights would sum to 1 by definition). We report results using this form of baseline model.
作为额外验证这些结果并非偶然出现的检查手段,我们还报告了基线(零)模型的结果。我们最初考虑使用两种形式的零模型:(1) 随机初始化权重的模型,以及 (2) 在随机打乱序列上训练的模型。然而在这两种情况下,数据集中没有任何序列产生的注意力权重能超过阈值 $\theta$。这表明分析中使用的高置信度注意力权重不可能偶然出现,但并未揭示具体分析过程的特性。因此我们实施了另一种随机化方案:在后处理步骤中随机打乱原始模型的注意力权重。具体而言,我们对每个注意力头中每个token的注意力权重序列进行置换。举例说明,假设原始模型在蛋白质序列 $x$(其中 $|x|=5$)的头 $h$ 中位置 $i$ 处产生的注意力权重为 (0.3, 0.2, 0.1, 0.4, 0.0),那么在零模型中,头 $h$ 序列 $x$ 位置 $i$ 的注意力权重将是这些权重的随机排列,例如 (0.2, 0.0, 0.4, 0.3, 0.1)。需要注意的是,这些仍然是有效的注意力权重,因为它们的总和为1(根据定义原始权重的总和即为1)。我们使用这种形式的基线模型报告结果。
B.3 PROBING METHODOLOGY
B.3 探测方法
Embedding probe. We probe the embedding vectors output from each layer using a linear probing classifier. For token-level probing tasks (binding sites, secondary structure) we feed each token’s output vector directly to the classifier. For token-pair probing tasks (contact map) we construct a pairwise feature vector by concatenating the element wise differences and products of the two tokens’ output vectors, following the $\mathrm{TAPE^{4}}$ implementation.
嵌入探针。我们使用线性探针分类器对每层输出的嵌入向量进行探测。对于token级探测任务(结合位点、二级结构),我们将每个token的输出向量直接输入分类器。对于token对探测任务(接触图),我们按照$\mathrm{TAPE^{4}}$实现方法,通过拼接两个token输出向量的逐元素差值和乘积来构建成对特征向量。
We use task-specific evaluation metrics for the probing classifier: for secondary structure prediction, we measure F1 score; for contact prediction, we measure precision $\mathcal{@}L/5$ , where $L$ is the length of the protein sequence, following standard practice (Moult et al., 2018); for binding site prediction, we measure precision $\mathcal{O}L/20$ , since approximately one in twenty amino acids in each sequence is a binding site ( $4.8%$ in the dataset).
我们针对探测分类器使用任务特定的评估指标:对于二级结构预测,我们测量F1分数;对于接触预测,我们测量精度$\mathcal{@}L/5$,其中$L$是蛋白质序列长度,遵循标准实践 (Moult et al., 2018);对于结合位点预测,我们测量精度$\mathcal{O}L/20$,因为每条序列中大约每20个氨基酸有一个结合位点(数据集中占比$4.8%$)。
Attention probe. Just as the attention weight $\alpha_ {i,j}$ is defined for a pair of amino acids $(i,j)$ , so is the contact property $f(i,j)$ , which returns true if amino acids $i$ and $j$ are in contact. Treating the attention weight as a feature of a token-pair $(i,j)$ , we can train a probing classifier that predicts the contact property based on this feature, thereby quantifying the attention mechanism’s knowledge of that property. In our multi-head setting, we treat the attention weights across all heads in a given layer as a feature vector, and use a probing classifier to assess the knowledge of a given property in the attention weights across the entire layer. As with the embedding probe, we measure performance of the probing classifier using precision $\mathcal{Q}L/5$ , where $L$ is the length of the protein sequence, following standard practice for contact prediction.
注意力探针。正如注意力权重 $\alpha_ {i,j}$ 是针对氨基酸对 $(i,j)$ 定义的,接触属性 $f(i,j)$ 也是如此,当氨基酸 $i$ 和 $j$ 接触时返回真值。将注意力权重视为token对 $(i,j)$ 的特征,我们可以训练一个探针分类器,基于该特征预测接触属性,从而量化注意力机制对该属性的认知。在多头注意力场景中,我们将给定层中所有注意力头的权重视为特征向量,并使用探针分类器评估整个注意力层对特定属性的认知能力。与嵌入探针类似,我们按照接触预测的标准做法,使用精度 $\mathcal{Q}L/5$ 衡量探针分类器的性能,其中 $L$ 为蛋白质序列长度。
B.4 DATASETS
B.4 数据集
We used two protein sequence datasets from the TAPE repository for the analysis: the ProteinNet dataset (AlQuraishi, 2019; Fox et al., 2013; Berman et al., 2000; Moult et al., 2018) and the Secondary Structure dataset (Rao et al., 2019; Berman et al., 2000; Moult et al., 2018; Klausen et al., 2019). The former was used for analysis of amino acids and contact maps, and the latter was used for analysis of secondary structure. We additionally created a third dataset for binding site and post-translational modification (PTM) analysis from the Secondary Structure dataset, which was augmented with binding site and PTM annotations obtained from the Protein Data Bank’s Web API.5 We excluded any sequences for which annotations were not available. The resulting dataset sizes are shown in Table 2. For the analysis of attention, a random subset of 5000 sequences from the training split of each dataset was used, as the analysis was purely evaluative. For training and evaluating the diagnostic classifier, the full training and validation splits were used.
我们使用了来自TAPE存储库的两个蛋白质序列数据集进行分析:ProteinNet数据集 (AlQuraishi, 2019; Fox et al., 2013; Berman et al., 2000; Moult et al., 2018) 和二级结构数据集 (Rao et al., 2019; Berman et al., 2000; Moult et al., 2018; Klausen et al., 2019) 。前者用于氨基酸和接触图分析,后者用于二级结构分析。我们还从二级结构数据集中创建了第三个数据集,用于结合位点和翻译后修饰 (PTM) 分析,该数据集通过从蛋白质数据库的Web API5获取的结合位点和PTM注释进行了增强。我们排除了任何没有可用注释的序列。最终的数据集大小如 表 2 所示。对于注意力分析,使用了每个数据集训练分割中的5000个随机序列子集,因为该分析纯粹是评估性的。对于训练和评估诊断分类器,使用了完整的训练和验证分割。
Table 2: Datasets used in analysis
| Dataset | Trainsize | Validationsize |
| ProteinNet | 25299 | 224 |
| Secondary Structure | 8678 | 2170 |
| Binding Sites / PTM | 5734 | 1418 |
表 2: 分析使用的数据集
| Dataset | Trainsize | Validationsize |
|---|---|---|
| ProteinNet | 25299 | 224 |
| Secondary Structure | 8678 | 2170 |
| Binding Sites / PTM | 5734 | 1418 |
C ADDITIONAL RESULTS OF ATTENTION ANALYSIS
C 注意力分析的附加结果
C.1 SECONDARY STRUCTURE
C.1 二级结构

Figure 8: Percentage of each head’s attention that is focused on Helix secondary structure.
图 8: 各注意力头(attention head)在螺旋二级结构(Helix secondary structure)上的注意力占比。

Figure 9: Percentage of each head’s attention that is focused on Strand secondary structure.
图 9: 各注意力头聚焦于Strand二级结构的注意力百分比。

Figure 10: Percentage of each head’s attention that is focused on Turn/Bend secondary structure.
图 10: 各注意力头聚焦于Turn/Bend二级结构的注意力百分比。

Figure 11: Top 10 heads (denoted by
图 11: 基于注意力与接触图对齐比例的各模型Top 10注意力头 (标记为<层>-<头>) [95%置信区间]。注意力比例与接触背景频率 (橙色虚线) 的差异具有统计学意义 (p<0.00001)。置信区间和检验均采用Bonferroni校正 (见附录B.2)。

Figure 12: Top-10 contact-aligned heads for null models. See Appendix B.2 for details.
图 12: 零样本模型的Top-10接触对齐注意力头。详见附录 B.2。

Figure 13: Top 10 heads (denoted by
图 13: 各模型中注意力聚焦于结合位点比例最高的前10个注意力头 (标记为<层数>-<头数>) [95% 置信区间]。注意力比例与结合位点背景频率 (橙色虚线) 的差异均具有统计学意义 (p<0.00001)。置信区间和检验均经过Bonferroni校正 (参见附录B.2)。

Figure 14: Top-10 heads most focused on binding sites for null models. See Appendix B.2 for details.
图 14: 零样本模型中最关注结合位点的Top-10注意力头。详见附录B.2。

Figure 15: Percentage of each head’s attention that is focused on post-translational modifications.
图 15: 各注意力头(attention head)聚焦于翻译后修饰(post-translational modifications)的注意力占比。

Figure 16: Top 10 heads (denoted by ${<l a y e r{>}}{-}{
图 16: 基于对PTM位置关注比例的各模型Top 10注意力头(表示为${<l a y e r{>}}{-}{

Figure 17: Top-10 heads most focused on PTMs for null models. See Appendix B.2 for details.
图 17: 零样本模型中最关注PTM的Top-10注意力头。详见附录B.2。
C.5 AMINO ACIDS
C.5 氨基酸

Figure 18: Percentage of each head’s attention that is focused on the given amino acid, averaged over a dataset (TapeBert).
图 18: 每个注意力头(attention head)对给定氨基酸的关注百分比,基于数据集(TapeBert)的平均值。

Figure 19: Percentage of each head’s attention that is focused on the given amino acid, averaged over a dataset (cont.)
图 19: 各注意力头对给定氨基酸的关注百分比(基于数据集平均值)(续)
Table 3: Amino acids and the corresponding maximally attentive heads in the standard and randomized versions of TapeBert. The differences between the attention percentages for TapeBert and the background frequencies of each amino acid are all statistically significant ( $p<0.00001)$ ) taking into account the Bonferroni correction. See Appendix B.2 for details. The bolded numbers represent the higher of the two values between the standard and random models. In all cases except for Glutamine, which was the amino acid with the lowest top attention proportion in the standard model (7.1), the standard TapeBert model has higher values than the randomized version.
| TapeBert | TapeBert-Random | ||||||
| Abbrev | Code | Name | Background % | Top Head | Attn % | Top Head | Attn % |
| Ala | A | Alanine | 7.9 | 12-11 | 25.5 | 11-12 | 12.1 |
| Arg | R | Arginine | 5.2 | 12-8 | 63.2 | 12-7 | 8.4 |
| Asn | N | Asparagine | 4.3 | 8-2 | 44.8 | 8-2 | 6.7 |
| Asp | D | Aspartic acid | 5.8 | 12-6 | 79.9 | 5-4 | 10.7 |
| Cys | C | Cysteine | 1.3 | 11-6 | 83.2 | 11-6 | 9.3 |
| Gln | Q | Glutamine | 3.8 | 11-7 | 7.1 | 12-1 | 9.2 |
| Glu | E | Glutamic acid | 6.9 | 11-7 | 16.2 | 11-4 | 11.8 |
| Gly | G | Glycine | 7.1 | 2-11 | 98.1 | 11-8 | 14.6 |
| His | H | Histidine | 2.7 | 9-10 | 56.7 | 11-6 | 5.4 |
| Ile | I | Isoleucine | 5.6 | 11-10 | 27.0 | 9-5 | 10.6 |
| Leu | L | Leucine | 9.4 | 2-12 | 44.1 | 12-11 | 13.9 |
| Lys | K | Lysine | 6.0 | 12-8 | 29.4 | 6-11 | 12.9 |
| Met | M | Methionine | 2.3 | 3-10 | 73.5 | 9-3 | 6.2 |
| Phe | F | Phenylalanine | 3.9 | 12-3 | 22.7 | 12-1 | 6.7 |
| Pro | P | Proline | 4.6 | 1-11 | 98.3 | 10-6 | 7.6 |
| Ser | S | Serine | 6.4 | 12-7 | 36.1 | 11-12 | 11.0 |
| Thr | T | Threonine | 5.4 | 12-7 | 19.0 | 10-4 | 9.0 |
| Trp | W | Tryptophan | 1.3 | 11-4 | 68.1 | 9-2 | 3.0 |
| Tyr | Y | Tyrosine | 3.4 | 12-3 | 51.6 | 12-11 | 6.6 |
| Val | V | Valine | 6.8 | 12-11 | 34.0 | 8-2 | 15.0 |
表 3: TapeBert标准版和随机版中各氨基酸及其对应最高注意力头。经Bonferroni校正后,TapeBert注意力百分比与各氨基酸背景频率之间的差异均具有统计学意义( $p<0.00001$ )。详情参见附录B.2。加粗数字表示标准模型与随机模型中较高值。除谷氨酰胺(标准模型中最高注意力比例最低的氨基酸,7.1)外,标准TapeBert模型在所有情况下均高于随机版本。
| 缩写 | 代码 | 名称 | 背景频率% | 最高注意力头 | 注意力% | 最高注意力头 | 注意力% |
|---|---|---|---|---|---|---|---|
| Ala | A | 丙氨酸 | 7.9 | 12-11 | 25.5 | 11-12 | 12.1 |
| Arg | R | 精氨酸 | 5.2 | 12-8 | 63.2 | 12-7 | 8.4 |
| Asn | N | 天冬酰胺 | 4.3 | 8-2 | 44.8 | 8-2 | 6.7 |
| Asp | D | 天冬氨酸 | 5.8 | 12-6 | 79.9 | 5-4 | 10.7 |
| Cys | C | 半胱氨酸 | 1.3 | 11-6 | 83.2 | 11-6 | 9.3 |
| Gln | Q | 谷氨酰胺 | 3.8 | 11-7 | 7.1 | 12-1 | 9.2 |
| Glu | E | 谷氨酸 | 6.9 | 11-7 | 16.2 | 11-4 | 11.8 |
| Gly | G | 甘氨酸 | 7.1 | 2-11 | 98.1 | 11-8 | 14.6 |
| His | H | 组氨酸 | 2.7 | 9-10 | 56.7 | 11-6 | 5.4 |
| Ile | I | 异亮氨酸 | 5.6 | 11-10 | 27.0 | 9-5 | 10.6 |
| Leu | L | 亮氨酸 | 9.4 | 2-12 | 44.1 | 12-11 | 13.9 |
| Lys | K | 赖氨酸 | 6.0 | 12-8 | 29.4 | 6-11 | 12.9 |
| Met | M | 甲硫氨酸 | 2.3 | 3-10 | 73.5 | 9-3 | 6.2 |
| Phe | F | 苯丙氨酸 | 3.9 | 12-3 | 22.7 | 12-1 | 6.7 |
| Pro | P | 脯氨酸 | 4.6 | 1-11 | 98.3 | 10-6 | 7.6 |
| Ser | S | 丝氨酸 | 6.4 | 12-7 | 36.1 | 11-12 | 11.0 |
| Thr | T | 苏氨酸 | 5.4 | 12-7 | 19.0 | 10-4 | 9.0 |
| Trp | W | 色氨酸 | 1.3 | 11-4 | 68.1 | 9-2 | 3.0 |
| Tyr | Y | 酪氨酸 | 3.4 | 12-3 | 51.6 | 12-11 | 6.6 |
| Val | V | 缬氨酸 | 6.8 | 12-11 | 34.0 | 8-2 | 15.0 |