Post-translational modification prediction via prompt-based fine-tuning of a GPT-2 model
基于GPT-2模型的提示微调实现翻译后修饰预测
Received: 11 March 2024
收到日期:2024年3月11日
Accepted: 29 July 2024
录用日期: 2024年7月29日
Published online: 07 August 2024
在线发布:2024年8月7日
Check for updates
检查更新
Palistha Shrestha 1,5, Jeevan Kandel 2,5, Hilal Tayara 3 & Kil To Chong 1,4
Palistha Shrestha 1,5, Jeevan Kandel 2,5, Hilal Tayara 3 & Kil To Chong 1,4
Post-translational modifications (PTMs) are pivotal in modulating protein functions and influencing cellular processes like signaling, localization, and degradation. The complexity of these biological interactions necessitates efficient predictive methodologies. In this work, we introduce PTMGPT2, an interpret able protein language model that utilizes prompt-based fine-tuning to improve its accuracy in precisely predicting PTMs. Drawing inspiration from recent advancements in GPT-based architectures, PTMGPT2 adopts unsupervised learning to identify PTMs. It utilizes a custom prompt to guide the model through the subtle linguistic patterns encoded in amino acid sequences, generating tokens indicative of PTM sites. To provide interpret ability, we visualize attention profiles from the model’s final decoder layer to elucidate sequence motifs essential for molecular recognition and analyze the effects of mutations at or near PTM sites to offer deeper insights into protein functionality. Comparative assessments reveal that PTMGPT2 outperforms existing methods across 19 PTM types, underscoring its potential in identifying disease associations and drug targets.
翻译后修饰 (PTM) 在调节蛋白质功能和影响细胞过程 (如信号传导、定位和降解) 中起着关键作用。这些生物相互作用的复杂性需要高效的预测方法。在这项工作中,我们引入了 PTMGPT2,这是一种可解释的蛋白质语言模型,它利用基于提示的微调来提高其精确预测 PTM 的准确性。受 GPT 架构最新进展的启发,PTMGPT2 采用无监督学习来识别 PTM。它利用自定义提示来引导模型理解氨基酸序列中编码的微妙语言模式,生成指示 PTM 位点的 Token。为了提供可解释性,我们可视化模型最后解码层的注意力分布,以阐明对分子识别至关重要的序列模体,并分析 PTM 位点处或附近突变的影响,以提供对蛋白质功能的更深入理解。比较评估表明,PTMGPT2 在 19 种 PTM 类型中优于现有方法,突显了其在识别疾病关联和药物靶点方面的潜力。
Proteins, the essential workhorses of the cell, are modulated by posttranslational modifications (PTM), a process vital for their optimal functioning. With over 400 known types of PTMs1, they enhance the functional spectrum of the proteome. Despite originating from a foundational set of approximately 20,000 protein-coding genes2, the dynamic realm of PTMs is estimated to expand the human proteome to over a million unique protein species. This diversity echoes the complexity inherent in human languages: individual amino acids assemble into’words’ to form functional’sentences’ or domains. This linguistic parallel extends to the dense, information-rich structure of both protein sequences and human languages. The advancing field of Natural Language Processing (NLP) not only unravels the intricacies of human communication but is also increasingly applied to decode the complex language of proteins. Our study leverages the transformative capabilities of generative pretrained transformers (GPT) based models, a cornerstone in NLP, to interpret and predict the complex landscape of PTMs, highlighting an intersection where computational linguistics meets molecular biology.
蛋白质作为细胞中不可或缺的主力军,其功能通过翻译后修饰 (PTM) 这一关键过程进行调控,以确保它们发挥最佳作用。目前已知的PTM类型超过400种1,它们拓展了蛋白质组的功能范围。尽管源自约20,000个蛋白质编码基因2这一基础集合,PTM的动态领域预计将人类蛋白质组扩展至超过百万种独特蛋白质变体。这种多样性映照了人类语言与生俱来的复杂性:单个氨基酸组装成"词汇",进而形成功能性的"句子"或结构域。这种语言学上的相似性延伸至蛋白质序列和人类语言共有的密集且信息丰富的结构特征。自然语言处理 (NLP) 领域的发展不仅揭示了人类交流的复杂性,也日益应用于解读蛋白质的复杂语言。我们的研究利用基于生成式预训练Transformer (GPT) 模型的变革性能力——这一NLP领域的基石技术,来解读和预测PTM的复杂景观,凸显了计算语言学与分子生物学交叉融合的前沿领域。
In the quest to predict PTM sites, the scientific community has predominantly relied on supervised methods, which have evolved significantly over the years3–5. These methods typically involve training algorithms on datasets where the modification status of each site is known, allowing the model to learn and predict modifications on new sequences. B.Trost and A.Kusalik6 initially focus on methods like Support Vector Machines and decision trees, which classify amino acid sequences based on geometric margins or hierarchical decision rules. Progressing towards more sophisticated approaches, Zhou, F. et al. 7 discuss the utilization of Convolutional Neural Networks and
在预测翻译后修饰 (PTM) 位点的研究中,科学界主要依赖于监督学习方法,这些方法经过多年发展已取得显著进步[3-5]。这类方法通常基于已知位点修饰状态的数据集训练算法,使模型能够学习并预测新序列的修饰情况。B.Trost 和 A.Kusalik[6] 最初聚焦于支持向量机和决策树等方法,这些方法通过几何间隔或分层决策规则对氨基酸序列进行分类。随着方法日益复杂,Zhou, F. 等人[7] 探讨了卷积神经网络的应用
Recurrent Neural Networks, adept at recognizing complex patterns and capturing temporal sequence dynamics. DeepSucc8 proposed a specific deep learning architecture for identifying succ in yl ation sites, indicative of the tailored application of deep learning in PTM prediction. Smith, L. M. and Kelleher, N. L9. highlight the challenges in data representation and quality in proteomics. Building upon these developments, Smith, D. et al. 10 further advance the field, presenting a deep learning-based approach that achieves high accuracy in PTM site prediction. Their method involves intricate neural network architectures optimized for analyzing protein sequences, signifying a refined integration of deep learning in protein sequence analysis.
循环神经网络 (Recurrent Neural Networks) ,擅长识别复杂模式并捕捉时间序列动态。DeepSucc8提出了一种特定的深度学习架构,用于识别琥珀酰化 (succinylation) 位点,这体现了深度学习在翻译后修饰预测中的定制化应用。Smith, L. M. 和 Kelleher, N. L [9] 强调了蛋白质组学中数据表示和质量的挑战。基于这些进展,Smith, D. 等人 [10] 进一步推动了该领域发展,提出了一种基于深度学习的方法,在翻译后修饰位点预测中实现了高精度。他们的方法涉及复杂的神经网络架构,这些架构经过优化以分析蛋白质序列,标志着深度学习在蛋白质序列分析中的精细化整合。
On the unsupervised learning front, methods like those developed by Chung, C. et al.11 have significantly advanced the field. Central to their algorithm is the clustering of similar features and the recognition of patterns indicative of PTM sites. Lee, Tzong-Yi, et al. 12 utilized distant sequence features in combination with Radial Basis Function Networks, an approach that effectively identifies ubiquitin conjugation sites by integrating non-local sequence information. However, the complexity of PTM processes, often characterized by subtle and context-dependent patterns, pose challenges to these methods. Despite their advancements, they often grapple with issues like data imbalance, where certain PTMs are underrepresented, and the dependency on high-quality annotated datasets. These approaches can be challenged by the intricate and subtle nature of PTM sites, potentially overlooking crucial biological details. This landscape of PTM site prediction is ripe for innovation through generative transformer models, particularly in the domain of unsupervised learning. Intrigued by this possibility, we explored the potential of generative transformers, exemplified by the GPT architecture, for predicting PTM sites.
在无监督学习方面,Chung, C.等人[11]开发的方法显著推动了该领域的发展。其算法的核心在于对相似特征进行聚类,并识别指示PTM位点的模式。Lee, Tzong-Yi等人[12]利用远距离序列特征结合径向基函数网络,通过整合非局部序列信息有效识别泛素结合位点。然而PTM过程通常具有细微且依赖上下文模式的特点,这种复杂性对这些方法构成了挑战。尽管取得了进展,这些方法仍需应对数据不平衡(某些PTM代表不足)和对高质量标注数据集的依赖等问题。PTM位点错综复杂的特性可能使这些方法难以捕捉关键的生物学细节。当前PTM位点预测领域正处于通过生成式Transformer模型实现突破的成熟阶段,特别是在无监督学习领域。受此前景吸引,我们探索了以GPT架构为代表的生成式Transformer在预测PTM位点方面的潜力。
Here, we introduce PTMGPT2, a suite of models capable of generating tokens that signify modified protein sequences, crucial for identifying PTM sites. At the core of this platform is PROTGPT213, an auto regressive transformer model. We have adapted PROTGPT2, utilizing it as a pre-trained model, and further fine-tuned it for the specific task of generating classification labels for a given PTM type. PTMGPT2 utilizes a decoder-only architecture, which eliminates the need for a task-specific classification head during training. Instead, the final layer of the decoder functions as a projection back to the vocabulary space, effectively generating the next possible token based on the learned patterns among tokens in the input prompt. When provided with a prompt, the model is faced with a protein sequence structured in a fillin-the-blank format. Impressively, even without any hyper parameter optimization procedures, our model has demonstrated an average $5.45%$ improvement in Matthews Correlation Coefficient (MCC) over all other competing methods. The webserver and models that underpin PTMGPT2 are available at https://nsclbio.jbnu.ac.kr/tools/ ptmgpt2. Given the critical role of PTM in elucidating the mechanisms of various biological processes, we believe PTMGPT2 represents a significant stride forward in the efficient prediction and analysis of protein sequences.
在此,我们推出PTMGPT2,这是一套能够生成表示修饰蛋白质序列的token的模型,对于识别PTM位点至关重要。该平台的核心是PROTGPT213,一种自回归Transformer模型。我们调整了PROTGPT2,将其用作预训练模型,并进一步针对生成特定PTM类型分类标签的任务进行了微调。PTMGPT2采用仅解码器架构,在训练过程中无需特定任务的分类头。相反,解码器的最后一层作为投影层返回词汇空间,基于输入提示中token之间的学习模式,有效地生成下一个可能的token。当提供提示时,模型面对的是以填空格式构建的蛋白质序列。令人印象深刻的是,即使没有任何超参数优化过程,我们的模型在马修斯相关系数 (MCC) 上比所有其他竞争方法平均提高了 $5.45%$ 。支持PTMGPT2的网站服务器和模型可在 https://nsclbio.jbnu.ac.kr/tools/ ptmgpt2 获取。鉴于PTM在阐明各种生物过程机制中的关键作用,我们相信PTMGPT2代表了在蛋白质序列高效预测和分析方面的重要进展。
Results
结果
PTMGPT2 implements a prompt-based approach for PTM prediction
PTMGPT2 采用基于提示的方法进行翻译后修饰 (PTM) 预测
We introduce an end-to-end deep learning framework depicted in Fig. 1, utilizing a GPT as the foundational model. Central to our approach is the prompt-based finetuning of the PROTGPT2 model in an unsupervised manner. This is achieved by utilizing informative prompts during training, enabling the model to generate accurate sequence labels. The design of these prompts is a critical aspect of our architecture, as they provide essential instructional input to the pretrained model, guiding its learning process. To enhance the explanatory power of these prompts, we have introduced four custom tokens to the pre-trained tokenizer, expanding its vocabulary size from 50,257 to 50,264. This modification is particularly significant due to the tokenizer’s reliance on the Byte Pair Encoding (BPE) algorithm 14. A notable consequence of this approach is that our model goes beyond annotating individual amino acid residues. Instead, it focuses on annotating variable-length protein sequence motifs. This strategy is pivotal as it ensures the preservation of evolutionary biological function ali ties, allowing for a more nuanced and biologically relevant interpretation of protein sequences.
我们引入了一个端到端的深度学习框架 (如图 1 所示) , 使用 GPT 作为基础模型。我们方法的核心是以无监督方式对 PROTGPT2 模型进行基于提示的微调。这是通过在训练期间使用信息性提示来实现的, 使模型能够生成准确的序列标签。这些提示的设计是我们架构的一个关键方面, 因为它们为预训练模型提供了必要的指令输入, 指导其学习过程。为了增强这些提示的解释能力, 我们向预训练的分词器引入了四个自定义 token, 将其词汇表大小从 50,257 扩展到 50,264。由于该分词器依赖于字节对编码 (BPE) 算法 [14], 这一修改尤为重要。这种方法的一个显著结果是, 我们的模型不仅能够注释单个氨基酸残基, 还能专注于注释可变长度的蛋白质序列 motif。该策略至关重要, 因为它确保了进化生物学功能特性的保留, 从而实现对蛋白质序列更细致且更具生物学意义的解读。
In the PTMGPT2 framework, we employ a prompt structure that incorporates four principal tokens. The first, designated as the ‘SEQUENCE:’ token, represents the specific protein sub sequence of interest. The second, known as the ‘LABEL:’ token, indicates whether the sub sequence is modified (‘POSITIVE’) or unmodified (‘NEGATIVE’). This token-driven prompt design forms the foundation for the fine-tuning process of the PTMGPT2 model, enabling it to accurately generate labels during inference. A key aspect of this model lies in its architectural foundation, which is based on GPT $\cdot2^{15}$ . This architecture is characterized by its exclusive use of decoder layers, with PTMGPT2 utilizing a total of 36 such layers, consistent with the pretrained model. This maintains architectural consistency while fine-tuning for our downstream task of PTM site prediction. Each of these layers is composed of masked self-attention mechanisms 16, which ensure that during the training phase, the protein sequence and custom tokens can be influenced only by their preceding tokens in the prompt. This is essential for maintaining the auto regressive property of the model. Such a method is fundamental for our model’s ability to accurately generate labels, as it helps preserve the chronological integrity of biological sequence data and its dependencies with custom tokens, ensuring that the predictions are biologically relevant.
在PTMGPT2框架中, 我们采用包含四个主要token的提示结构。第一个是 "SEQUENCE:" token, 代表目标蛋白质亚序列。第二个是 "LABEL:" token, 指示该亚序列是否被修饰 ("POSITIVE") 或未被修饰 ("NEGATIVE") 。这种基于token的提示设计构成了PTMGPT2模型微调过程的基础, 使其能够在推理过程中准确生成标签。该模型的一个关键特性在于其架构基础基于GPT $\cdot2^{15}$ 。该架构的特点是仅使用解码器层, PTMGPT2共使用36个这样的层, 与预训练模型保持一致。这在对我们的PTM位点预测下游任务进行微调时保持了架构一致性。每个层都由掩码自注意力机制16组成, 确保在训练阶段, 蛋白质序列和自定义token仅受提示中前序token的影响。这对于保持模型的自回归特性至关重要。这种方法对我们模型准确生成标签的能力至关重要, 因为它有助于保持生物序列数据的时间完整性及其与自定义token的依赖关系, 确保预测结果具有生物学意义。
A key distinction in our approach lies in the methodology we employed for prompt based fine-tuning during the training and inference phases of PTMGPT2. During the training phase, PTMGPT2 is engaged in an unsupervised learning process. This approach involves feeding the model with input prompts and training it to output the same prompt, thereby facilitating the learning of token relationships and context within the prompts themselves. This process enables the model to generate the next token based on the patterns learned during training between protein sub sequences and their corresponding labels. The approach shifts during the inference phase, where the prompts are modified by removing the ‘POSITIVE’ and ‘NEGATIVE’ tokens, effectively turning these prompts into a fill-in-the-blank exercise for the model. This strategic masking triggers PTMGPT2 to generate the labels independently, based on the patterns and associations it learned during the training phase. An essential aspect of our prompt structure is the consistent inclusion of the ‘<start of text $>^{\prime}$ and ‘<endoftext $>^{\prime}$ tokens. These tokens are integral to our prompts, signifying the beginning and end of the prompt helping the model to contextual ize the input more effectively. This interplay of training techniques and strategic prompt structuring enables PTMGPT2 to achieve high prediction accuracy and efficiency. Such an approach sets PTMGPT2 apart as an advanced tool for protein sequence analysis, particularly in predicting PTMs.
我们方法的一个关键区别在于PTMGPT2训练和推理阶段采用的基于提示的微调方法。在训练阶段,PTMGPT2进行无监督学习,通过输入提示训练模型输出相同提示,从而学习token关系及提示内部的上下文关联。这使得模型能够根据训练中学到的蛋白质子序列与其对应标签之间的模式来生成下一个token。在推理阶段,该方法发生转变:通过移除"POSITIVE"和"NEGATIVE"token将提示转化为填空任务,这种策略性掩码触发PTMGPT2基于训练阶段学习的模式关联独立生成标签。我们提示结构的重要特点是始终包含<start of text \$>^{\prime}\$和<endoftext \$>^{\prime}\$token,这些标记提示的起止位置,帮助模型更有效地理解输入上下文。这种训练技术与策略性提示结构的结合,使PTMGPT2实现了高预测精度和效率,使其成为蛋白质序列分析(尤其是翻译后修饰预测)的先进工具。
Effect of prompt design and fine-tuning on PTMGPT2 performance
提示设计和微调对PTMGPT2性能的影响
We designed five prompts with custom tokens (‘SEQUENCE:’, ‘LABEL:’, ‘POSITIVE’, and ‘NEGATIVE’) to identify the most efficient one for capturing complexity, allowing PTMGPT2 to learn and process specific sequence segments for more meaningful representations. Initially, we crafted a prompt that integrates all custom tokens with a 21-length protein sub sequence. Subsequent explorations were conducted with 51-length sub sequence and 21-length sub sequence split into groups of k-mers, with and without the custom tokens. Considering that the pretrained model was originally trained solely on protein sequences, we
我们设计了五个包含自定义标记 ( 'SEQUENCE:', 'LABEL:', 'POSITIVE', 'NEGATIVE' ) 的提示方案, 以确定最能有效捕捉复杂性的方案, 使 PTMGPT2 能够学习并处理特定序列片段, 从而获得更有意义的表征。最初, 我们构建了一个整合所有自定义标记与 21 长度蛋白质子序列的提示方案。后续探索采用 51 长度子序列, 以及将 21 长度子序列拆分为 k-mer 组的形式, 分别测试了包含与不包含自定义标记的情况。考虑到预训练模型最初仅基于蛋白质序列进行训练, 我们
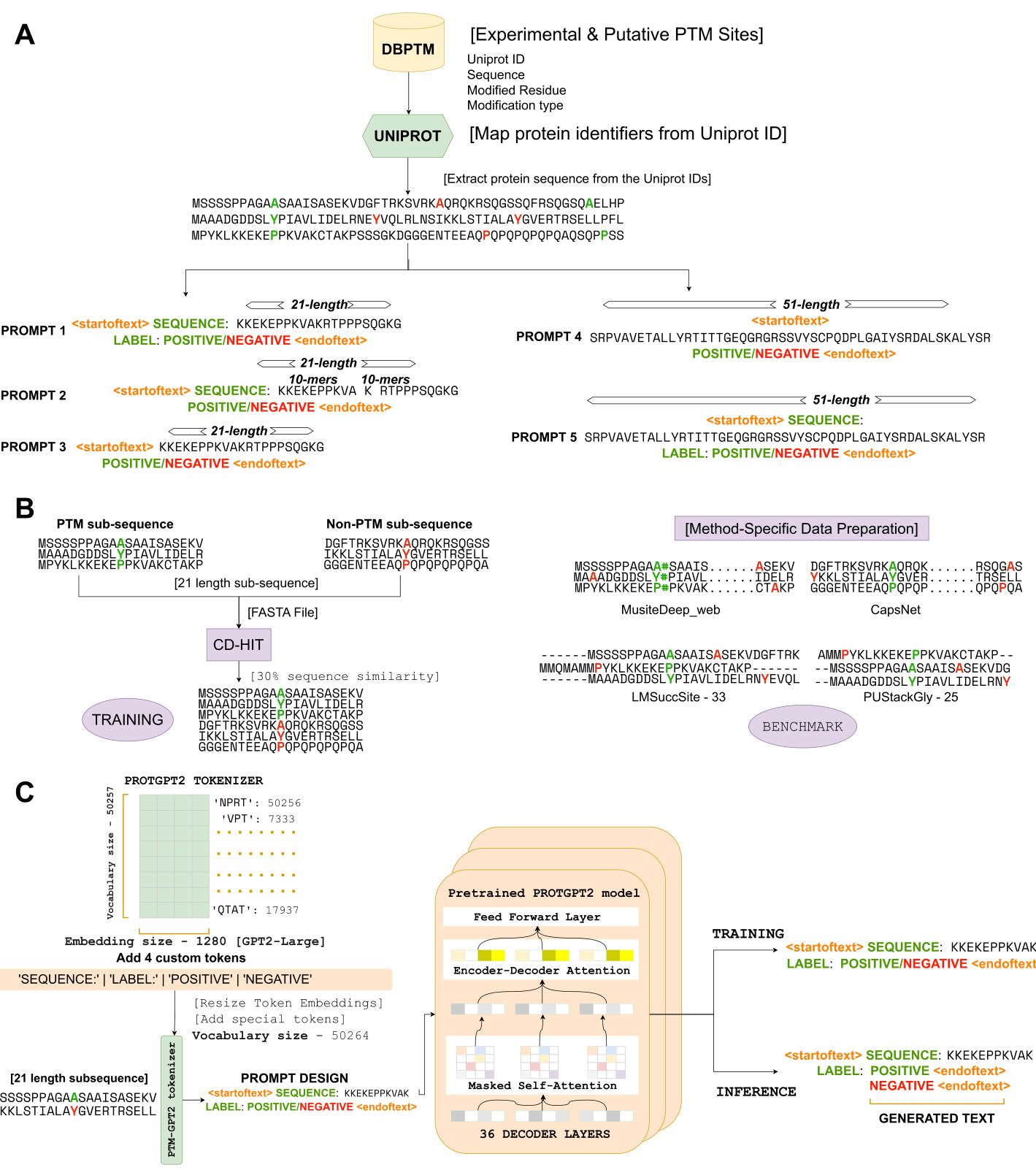
Fig. 1 | Schematic representation of the PTMGPT2 framework. A Preparation of inputs for PTMGPT2, detailing the extraction of protein sequences from Uniprot and the generation of five distinct prompt designs. B Method-specific data preparation process for benchmark, depicting both modified and unmodified subsequence extraction, followed by the creation of a training dataset using CD-HIT for
图 1 | PTMGPT2 框架示意图。A PTMGPT2 的输入数据准备流程,详细说明从 Uniprot 提取蛋白质序列及生成五种不同提示设计的过程。B 基准测试的特定方法数据准备流程,展示修饰与未修饰子序列的提取步骤,随后通过 CD-HIT 构建训练数据集。
$30%$ sequence similarity C Architecture of the PTMGPT2 model and the training and inference processes. It highlights the integration of custom tokens into the tokenizer, the resizing of token embeddings, and the subsequent prompt design utilized during training and inference to generate predictions.
$30%$ 序列相似性 C PTMGPT2 模型架构及训练与推理流程。该图展示了将自定义 token 集成至分词器、调整 token 嵌入维度,以及在训练和推理过程中用于生成预测的后续提示设计。
fine-tuned it with prompts both with and without the tokens to ascertain their actual contribution to improving PTM predictions.
通过使用包含和不包含这些token的提示进行微调, 以确定它们对改进PTM预测的实际贡献。
Upon fine-tuning PTMGPT2 with training datasets for arginine(R) methyl ation and tyrosine(Y) phosphor yl ation, it became evident that the prompt containing the 21-length sub sequence and the four custom tokens yielded the best results in generating accurate labels, as shown in Table 1. For methyl ation (R), the MCC, F1 Score, precision, and recall were reported as 80.51, 81.32, 95.14, and 71.01, respectively. Similarly, for phosphor yl ation (Y), the MCC, F1 Score, precision, and recall were
在使用精氨酸 (R) 甲基化和酪氨酸 (Y) 磷酸化训练数据集对 PTMGPT2 进行微调后,我们发现包含 21 长度子序列和四个自定义 token 的提示在生成准确标签方面效果最佳,如表 1 所示。对于甲基化 (R) ,其 MCC、F1 分数、精确度和召回率分别为 80.51、81.32、95.14 和 71.01。同样地,对于磷酸化 (Y) ,其 MCC、F1 分数、精确度和召回率分别为
48.83, 46.98, 30.95, and 97.51, respectively. So, for all the experiments, we used the 21-length sequence with custom tokens. The inclusion of ‘SEQUENCE:’ and ‘LABEL:’ tokens provided clear contextual cues to the model, allowing it to understand the structure of the input and the expected output format. This helped the model differentiate between the sequence data and the classification labels, leading to better learning and prediction accuracy. The 21-length sub sequence was an ideal size for the model to capture the necessary information without being too short to miss important context or too long to introduce noise. By framing the task clearly with the ‘SEQUENCE:’ and ‘LABEL:’ tokens, the model faced less ambiguity in generating predictions, which can be particularly beneficial for complex tasks such as PTM site prediction.
分别为48.83、46.98、30.95和97.51。因此在所有实验中, 我们采用了包含自定义token的21长度序列。"SEQUENCE:"和"LABEL:" token的加入为模型提供了清晰的上下文线索, 使其能够理解输入数据的结构和预期输出格式。这有助于模型区分序列数据和分类标签, 从而提升学习效果和预测准确性。21长度子序列是理想尺寸, 既能确保模型捕获必要信息, 又避免了因过短而遗漏关键上下文或因过长而引入噪声。通过"SEQUENCE:"和"LABEL:" token明确界定任务, 模型在生成预测时面临的模糊性显著降低, 这对于像PTM位点预测这类复杂任务尤为有利。
Table 1 | Benchmark results of PTMGPT2 after fine-tuning for optimal prompt selection
| PTM | Prompt | MCC F1Score | Precision | Recall |
| Methylation(R) | 21-lengthw/tokens[Proposed] 80.51 | 81.32 | 95.14 | 71.01 |
| 21-lengthk-merw/otokens 77.16 | 78.53 | 94.98 | 65.03 | |
| 51-lengthw/otokens | 58.25 63.37 | 85.02 | 33.56 | |
| 51-lengthw/tokens | 60.85 66.98 | 89.34 | 45.77 | |
| Phosphorylation(Y) | 21-lengthw/tokens[Proposed] | 48.83 46.98 | 30.95 | 97.51 |
| 21-lengthw/otokens | 45.27 44.07 | 30.04 | 90.47 | |
| 51-lengthw/otokens | 27.48 31.25 | 20.32 | 67.56 | |
| 51-lengthw/tokens | 31.01 32.76 | 20.71 | 78.37 |
Top values are represented in bold.
表 1 | 微调后 PTMGPT2 最优提示选择的基准测试结果
| PTM | Prompt | MCC F1Score | Precision | Recall |
|---|---|---|---|---|
| Methylation (R) | 21-lengthw/tokens [Proposed] | 80.51 | 81.32 | 95.14 |
| 21-lengthk-merw/otokens | 77.16 | 78.53 | 94.98 | |
| 51-lengthw/otokens | 58.25 | 63.37 | 85.02 | |
| 51-lengthw/tokens | 60.85 | 66.98 | 89.34 | |
| Phosphorylation (Y) | 21-lengthw/tokens [Proposed] | 48.83 | 46.98 | 30.95 |
| 21-lengthw/otokens | 45.27 | 44.07 | 30.04 | |
| 51-lengthw/otokens | 27.48 | 31.25 | 20.32 | |
| 51-lengthw/tokens | 31.01 | 32.76 | 20.71 |
最高值以粗体表示。
Comparative benchmark analysis reveals PTMGPT2’s dominance
比较基准分析显示 PTMGPT2 占据主导地位
To validate PTMGPT2’s performance, benchmarking against a database that encompasses a broad spectrum of experimentally verified PTMs and annotates potential PTMs for all UniProt17 entries was imperative. Accordingly, we chose the DBPTM database18 for its extensive collection of benchmark datasets, tailored for distinct types of PTMs. The inclusion of highly imbalanced datasets from DBPTM proved to be particularly advantageous, as it enabled a precise evaluation of PTMGPT2s ability to identify unmodified amino acid residues. This capability is crucial, considering that the majority of residues in a protein sequence typically remain unmodified. For a thorough assessment, we sourced 19 distinct benchmarking datasets from DBPTM, each containing a minimum of 500 data points corresponding to a specific PTM type.
为验证 PTMGPT2 的性能,必须使用包含广泛实验验证的翻译后修饰 (PTM) 并注释所有 UniProt17 条目潜在 PTM 的数据库进行基准测试。因此,我们选择 DBPTM 数据库 [18] ,其专为不同类型的 PTM 定制了广泛的基准数据集。DBPTM 中高度不平衡数据集的纳入被证明特别有利,因为它能精确评估 PTMGPT2 识别未修饰氨基酸残基的能力。考虑到蛋白质序列中大多数残基通常保持未修饰状态,这一能力至关重要。为进行全面评估,我们从 DBPTM 获取了 19 个不同的基准数据集,每个数据集至少包含 500 个对应特定 PTM 类型的数据点。
Our comparative analysis underscores PTMGPT2’s capability in predicting a variety of PTMs, marking substantial improvements when benchmarked against established methodologies using the MCC as the metric as shown in Table 2. For instance, in the case of lysine(K) succinylation, Succ-PTMGPT2 achieved a notable $7.94%$ improvement over LM-SuccSite. In the case of lysine(K) sumo yl ation, SumoyPTMGPT2 surpassed GPS Sumo by $5.91%$ . The trend continued with N-linked g lycos yl ation on asparagine(N), where N-linked-PTMGPT2 outperformed Musite-Web by $5.62%$ . RMethyl-PTMGPT2, targeting arginine(R) methyl ation, surpassed Musite-Web by $12.74%$ . Even in scenarios with marginal gains, such as lysine(K) acetyl ation where KAcetyl-PTMGPT2 edged out Musite-web by $0.46%$ , PTMGPT2 maintained its lead. PTMGPT2 exhibited robust performance for lysine(K) ubiquitin ation, surpassing Musite-Web by $5.01%$ . It achieved a $9.08%$ higher accuracy in predicting O-linked g lycos yl ation on serine(S) and threonine(T) residues. For cysteine(C) S-nitro syl ation, the model outperformed PresSNO by $4.09%$ . In lysine(K) mal on yl ation, PTMGPT2’s accuracy exceeded that of DL-Malosite by $3.25%$ , and for lysine(K) methyl ation, it achieved $2.47%$ higher accuracy than MethylSite. Although PhosphoST-PTMGPT2’s performance in serine-threonine (S, T) phosphor yl ation prediction was $16.37%$ , lower than Musite-Web, it excelled in tyrosine(Y) phosphor yl ation with an accuracy of $48.83%$ , which was notably higher than Musite-Web’s $40.83%$ and Capsnet’s $43.85%$ . In the case of cysteine (C) glut at hi on yl ation and lysine (K) glut ary la tion, Glut at hi oPT MG PT 2 and Glutary-PTMGPT2 exhibited improvements of $7.51%$ and $6.48%$ over DeepGSH and ProtTrans-Glutar, respectively. In the case of valine (V) amidation and cysteine (C) spalm i to yl ation, Ami-PTMGPT2 and Palm-PTMGPT2 surpassed prAS and CapsNet by $4.78%$ and $1.56%$ , respectively. Similarly, in the cases of proline (P) hydroxyl ation, lysine (K) hydroxyl ation, and lysine (K)
我们的比较分析凸显了PTMGPT2在预测多种翻译后修饰方面的能力, 如表2所示, 以MCC作为评估指标时, 相较于已有方法取得了显著提升。例如在赖氨酸(K)琥珀酰化预测中, Succ-PTMGPT2较LM-SuccSite实现了7.94%的显著提升。在赖氨酸(K)苏素化预测中, SumoyPTMGPT2以5.91%的优势超越GPS-Sumo。这一优势在天冬酰胺(N)的N-连接糖基化预测中得以延续, N-linked-PTMGPT2以5.62%的准确率超越Musite-Web。针对精氨酸(R)甲基化的RMethyl-PTMGPT2则以12.74%的幅度领先Musite-Web。即使在提升幅度较小的场景中, 如赖氨酸(K)乙酰化预测中KAcetyl-PTMGPT2仅以0.46%的微弱优势超越Musite-Web, PTMGPT2仍保持领先地位。
PTMGPT2在赖氨酸(K)泛素化预测中展现出稳健性能, 以5.01%的优势超越Musite-Web。在丝氨酸(S)和苏氨酸(T)残基的O-连接糖基化预测中, 其准确率提升达9.08%。对于半胱氨酸(C)S-亚硝基化预测, 该模型以4.09%的优势超越PresSNO。在赖氨酸(K)丙二酰化预测中, PTMGPT2的准确率较DL-Malosite提升3.25%, 在赖氨酸(K)甲基化预测中较MethylSite提升2.47%。尽管PhosphoST-PTMGPT2在丝氨酸-苏氨酸(S,T)磷酸化预测中的表现较Musite-Web低16.37%, 但在酪氨酸(Y)磷酸化预测中以48.83%的准确率显著优于Musite-Web的40.83%和Capsnet的43.85%。
在半胱氨酸(C)谷胱甘肽化和赖氨酸(K)戊二酰化预测中, GlutathioPTMGPT2和Glutary-PTMGPT2分别较DeepGSH和ProtTrans-Glutar提升7.51%和6.48%。在缬氨酸(V)酰胺化和半胱氨酸(C)棕榈酰化预测中, Ami-PTMGPT2和Palm-PTMGPT2分别以4.78%和1.56%的优势超越prAS和CapsNet。类似地, 在脯氨酸(P)羟基化、赖氨酸(K)羟基化等预测任务中...
for myl ation, PTMGPT2 achieved superior performance over CapsNet by $11.02%$ , $7.58%$ , and $4.39%$ , respectively. Collectively, these results demonstrate the significant progress made by PTMGPT2 in advancing the precision of PTM site prediction, thereby solidifying its place as a leading tool in proteomics research.
在我的评估中,PTMGPT2相较于CapsNet分别实现了11.02%、7.58%和4.39%的性能提升。这些结果共同证明了PTMGPT2在推进PTM位点预测精度方面取得的显著进展,从而巩固了其在蛋白质组学研究中的领先工具地位。
PTMGPT2 captures sequence-label dependencies through an attention-driven interpret able framework
PTMGPT2通过注意力驱动的可解释框架捕捉序列标签依赖关系
To enable PTMGPT2 to identify critical sequence determinants essential for protein modifications, we designed a framework depicted in Fig. 2A that processes protein sequences to extract attention scores from the model’s last decoder layer. The attention mechanism is pivotal as it selectively weighs the importance of different segments of the input sequence during prediction. Particularly, the extracted attention scores from the final layer provided a granular view of the model’s focus across the input sequence. By aggregating the attention across 20 attention heads (AH) for each position in the sequence, PTMGPT2 revealed which amino acids or motifs the model deemed crucial in relation to the ‘POSITIVE’ token. The Position Specific Probability Matrix $(\mathsf{P S P M})^{19}$ , characterized by rows representing sequence positions and columns indicating amino acids, was a key output of this analysis. It sheds light on the proportional representation of each amino acid in the sequences, as weighted by the attention scores. PTMGPT2 thus offers a refined view of the probabilistic distribution of amino acid occurrences, revealing key patterns and preferences in amino acid positioning.
为使PTMGPT2能够识别蛋白质修饰所必需的关键序列决定因素,我们设计了图2A所示的框架,该框架处理蛋白质序列以从模型的最后一个解码器层提取注意力分数。注意力机制至关重要,因为它能在预测过程中选择性权衡输入序列不同片段的重要性。特别地,从最终层提取的注意力分数提供了模型在输入序列上关注焦度的细粒度视图。通过聚合序列中每个位置20个注意力头(AH)的注意力权重,PTMGPT2揭示了模型认为与"POSITIVE"标记相关的关键氨基酸或基序。位置特异性概率矩阵$(\mathsf{P S P M})^{19}$是该分析的关键输出,其行代表序列位置,列表示氨基酸。该矩阵通过注意力分数加权,阐明了各氨基酸在序列中的比例分布。PTMGPT2由此提供了氨基酸出现概率分布的精细化视图,揭示了氨基酸定位的关键模式与偏好。
Motifs $\mathbf{K}^{**}A^{*}A$ and $C^{**}K$ were identified in AH 10, while motifs $K^{\ast\ast\ast}\mathsf{K}^{\ast\ast\ast\ast}\mathsf{A}$ , $^{*}\mathrm{KH^{*}}$ , and ${\mathsf{K}}^{***}{\mathsf{K}}$ were detected in AH 7. In AH 19, motifs $\mathsf{K}^{*}\mathsf{C}$ and $\mathtt{C}^{*}\mathtt{K}$ motifs were observed, and the $\mathbf{*}\mathbf{G}\mathbf{K}^{*}$ motif was found in AH 15. Furthermore, motifs ${}^{*}\mathbf{E}\mathbf{K}^{*}$ and $\mathbf{K}\mathbf{L}^{**}\mathbf{E}\mathbf{R}$ were identified in AH 5, motifs $\mathsf{H}^{***}\mathsf{K}$ , $\mathbf{D}^{**}\mathbf{K},$ and ${}^{*}\mathsf{F K}^{*}$ were detected in $\mathsf{A H4}.$ The $\mathbf{A}^{**}\mathbf{K}$ motif was observed in AH13, $\mathbf{A}^{*}\mathbf{K}$ motif in AH2, and ${}^{*}\mathbf{KM}^{*}$ motif in AH 11. To validate the predictions made by PTMGPT2 for lysine (K) acetyl ation, as shown in Fig. 2B, we compared these with motifs identified in prior research that has undergone experimental validation. Expanding our analysis to protein kinase domains, we visualized motifs for the CMGC and AGC kinase families, as shown in Fig. 3A, B. Additionally, the motifs for the CAMK kinase family and general protein kinases are shown in Fig. 4A, B, respectively. The CMGC kinase family named after its main members, CDKs (cyclin-dependent kinases), MAPKs (mitogen-acti- vated protein kinases), GSKs (glycogen synthase kinases), and CDK-like kinases is involved in cell cycle regulation, signal transduction, and cellular differentiation 20. PTMGPT2 identified the common motif ${}^{*}{\mathsf{P}}^{*}{\mathsf{S P}}^{*}$ (Proline at positions $^{-2}$ and $^{+1}$ from the phosphor y late d serine residue) in this family. The AGC kinase family, comprising key serine/ threonine protein kinases such as PKA (protein kinase A), PKG (protein kinase G), and PKC (protein kinase C), plays a critical role in regulating metabolism, growth, proliferation, and survival21. The predicted common motif in this family was $\mathbf{R}^{**}\mathbf{S}\mathbf{L}$ (Arginine at position $^{-2}$ and leucine at position $^{+1}$ from either a phosphor y late d serine or threonine). The
在AH 10中识别到基序 $K^{**}\mathbf{A}^{*}\mathbf{A}$ 和 $\mathtt{C}^{**}\mathtt{K}$,而在AH 7中检测到基序 $\mathsf{K}^{\ast\ast\ast}\mathsf{K}^{\ast\ast\ast\ast}\mathsf{A}$、$^{*}\mathrm{KH^{*}}$ 和 ${\mathsf{K}}^{***}{\mathsf{K}}$。在AH 19中观察到基序 $\mathsf{K}^{*}\mathsf{C}$ 和 $\mathtt{C}^{*}\mathtt{K}$,在AH 15中发现基序 $\mathbf{*}\mathbf{G}\mathbf{K}^{*}$。此外,在AH 5中识别到基序 ${}^{*}\mathbf{E}\mathbf{K}^{*}$ 和 $\mathbf{K}\mathbf{L}^{**}\mathbf{E}\mathbf{R}$,在 $\mathsf{A H4}$ 中检测到基序 $\mathsf{H}^{***}\mathsf{K}$、$\mathbf{D}^{**}\mathbf{K},$ 和 ${}^{*}\mathsf{F K}^{*}$。在AH13中观察到基序 $\mathbf{A}^{**}\mathbf{K}$,在AH2中观察到基序 $\mathbf{A}^{*}\mathbf{K}$,在AH 11中观察到基序 ${}^{*}\mathbf{KM}^{*}$。为验证PTMGPT2对赖氨酸 (K) 乙酰化的预测(如图2B所示),我们将其与既往经过实验验证的研究中已识别的基序进行比较。将分析扩展至蛋白激酶结构域,我们可视化了CMGC和AGC激酶家族的基序(如图3A、B所示)。此外,CAMK激酶家族和通用蛋白激酶的基序分别显示在图4A、B中。CMGC激酶家族以其主要成员命名,包括CDKs(细胞周期蛋白依赖性激酶)、MAPKs(丝裂原活化蛋白激酶)、GSKs(糖原合成酶激酶)和CDK样激酶,参与细胞周期调控、信号转导和细胞分化[20]。PTMGPT2在该家族中识别出常见基序 ${}^{*}{\mathsf{P}}^{*}{\mathsf{S P}}^{*}$(在磷酸化丝氨酸残基的 $^{-2}$ 和 $^{+1}$ 位置为脯氨酸)。AGC激酶家族包含关键的丝氨酸/苏氨酸蛋白激酶,如PKA(蛋白激酶A)、PKG(蛋白激酶G)和PKC(蛋白激酶C),在调节代谢、生长、增殖和存活中起关键作用[21]。该家族预测的常见基序为 $\mathbf{R}^{**}\mathbf{S}\mathbf{L}$(在磷酸化丝氨酸或苏氨酸的 $^{-2}$ 位置为精氨酸,$^{+1}$ 位置为亮氨酸)。
Table 2 | Benchmark dataset results
| PTM | Model | MCC | F1Score | Precision | Recall |
| Succinylation (K) | LM-SuccSite34 | 43.94 | 62.26 | 53.05 | 45.34 |
| Psuc-EDBAM35 | 43.15 | 58.23 | 51.03 | 47.8 | |
| SuccinSite36 | 15.87 | 34.47 | 64.95 | 23.46 | |
| Deep SuccinylSite37 | 39.77 | 59.4 | 70.51 | 48.32 | |
| Deep-KSuccSite38 | 36.88 | 55.89 | 47.41 | 48.06 | |
| Succ-PTMGPT2 | 51.88 | 60.74 | 79.15 | 49.27 | |
| Sumoylation (K) | Musite-Web39 | 42.84 | 41.73 | 63.96 | 27.21 |
| CapsNet40 | 37.05 | 33.35 | 68.75 | 20.45 | |
| ResSum041 | 21.07 | 25.59 | 15.56 | 61.97 | |
| GPS Sum042 | 61.02 | 65.76 | 70.38 | 25.67 | |
| Sumoy-PTMGPT2 | 66.93 | 69.21 | 75.87 | 63.63 | |
| N-linked Glycosylation (N) | Musite-Web39 | 65.30 | 68.56 | 61.42 | 87.08 |
| CapsNet40 | 50.38 | 56.18 | 66.04 | 45.65 | |
| LMNglyPred43 | 39.34 | 35.17 | |||
| N-linked-PTMGPT2 | 70.92 | 74.21 | 57.49 64.35 | 14.58 87.65 | |
| Methyl Arginine (R) | Musite-Web39 | 67.77 | 68.99 | 85.41 | 57.86 |
| CapsNet40 | 65.39 | 65.12 | 90.03 | 51.08 | |
| DeepRMethylSite44 | 34.02 | 52.49 | 79.89 | 39.08 | |
| PRmePred45 | 53.76 | 54.33 | 76.45 | 36.22 | |
| CNNArginineMe46 | 34.55 | 36.98 | 97.42 | 22.82 | |
| Lysine Acetylation (K) | RMethyl-PTMGPT2 | 80.51 | 81.32 | 95.14 | 71.01 |
| Musite-Web39 | 21.63 | 36.14 | 94.86 | 22.32 | |
| CapsNet40 | 21.62 | 36.54 | 93.46 | ||
| GPS-PAIL47 | 12.35 | 16.14 | 72.63 | 22.71 9.07 | |
| Ubiquitination (K) | KAcetyl-PTMGPT2 | 22.09 | 40.51 | 96.06 | 25.67 |
| Musite-Web39 | 26.24 | 27.67 | 75.67 | 16.93 | |
| DL-Ubiq48 | 10.91 | 33.37 | |||
| Ubiq-PTMGPT2 | 31.25 | 35.74 | 36.67 80.46 | 23.76 22.97 | |
| O-linked-Glycosylation (S,T) | Musite-Web39 | 49.89 | 50.29 | 61.64 | 40.38 |
| OGlyThr49 | 41.6 | 50.71 | 52.82 | 60.78 | |
| GlyCopp50 | 41.05 | 14.38 | |||
| O-linked-PTMGPT2 | 58.97 | 61.80 | 50.85 | 46.67 65.14 | |
| S-Nitrosylation (C) | PCysMod51 | 48.53 | 66.67 | 58.79 62.50 | 71.42 |
| DeepNitro52 | 79.55 | 45.45 | |||
| PresSNO53 | 59.45 | 62.50 74.21 | 72.72 | ||
| pIMSNOSite54 | 69.52 | 84.19 | |||
| SNitro-PTMGPT2 | 55.91 | 17.60 | 14.74 | 21.82 | |
| Malonylation (K) | DL-Malosite55 | 73.61 69.78 | 80.49 77.12 | 84.30 | 77.01 81.94 |
| Methyl Lysine (K) | Maloy-PTMGPT2 | 73.03 | 78.14 | 72.83 82.95 | 73.85 |
| Musite-Web39 | 14.97 | 10.13 | 72.44 | 5.44 | |
| MethylSite56 | 38.60 | 35.54 | 39.41 | 66.66 | |
| KMethyl-PTMGPT2 | 41.07 | 36.35 | 88.68 | 22.86 | |
| Phosphorylation (S, T) | Musite-Web39 | 17.73 | 12.22 | 6.67 | 72.74 |
| CapsNet40 | 6.71 | 71.94 | |||
| PhosphoST-PTMGPT2 | 16.23 16.37 | 12.28 15.35 | 9.14 | 46.97 | |
| Musite-Web39 | 40.83 | 43.03 | 29.82 | 77.27 | |
| Phosphorylation (Y) | CapsNet40 | 43.85 | 46.61 | 36.58 | 68.18 |
| PhosphoY-PTMGPT2 | 48.83 | 46.98 | 30.95 | 97.51 | |
| DeepGSH57 | 70.77 | 75.85 | |||
| Glutathio-PTMGPT2 | 74.23 | 75.28 | |||
| ProtTrans-Glutar58 | 78.28 | 82.37 | 90.07 | 75.89 | |
| Glutarylation (K) | 62.99 | 59.45 | 68.09 | 79.16 | |
| Glutary-PTMGPT2 PrAS59 | 69.47 76.00 | 73.78 NR | 71.02 NR | 76.76 81.2 | |
| Ami-PTMGPT2 | 80.78 | 76.59 | 66.67 | 86.76 | |
| Musite-Web39 | 35.69 | 47.82 | 79.78 | 49.45 |
表 2 | 基准数据集结果
| PTM | 模型 | MCC | F1分数 | 精确率 | 召回率 |
|---|---|---|---|---|---|
| 琥珀酰化 (K) | |||||
| LM-SuccSite[34] | 43.94 | 62.26 | 53.05 | 45.34 | |
| Psuc-EDBAM[35] | 43.15 | 58.23 | 51.03 | 47.8 | |
| SuccinSite[36] | 15.87 | 34.47 | 64.95 | 23.46 | |
| Deep SuccinylSite[37] | 39.77 | 59.4 | 70.51 | 48.32 | |
| Deep-KSuccSite[38] | 36.88 | 55.89 | 47.41 | 48.06 | |
| Succ-PTMGPT2 | 51.88 | 60.74 | 79.15 | 49.27 | |
| 苏素化 (K) | |||||
| Musite-Web[39] | 42.84 | 41.73 | 63.96 | 27.21 | |
| CapsNet[40] | 37.05 | 33.35 | 68.75 | 20.45 | |
| ResSumo[41] | 21.07 | 25.59 | 15.56 | 61.97 | |
| GPS Sumo[42] | 61.02 | 65.76 | 70.38 | 25.67 | |
| Sumoy-PTMGPT2 | 66.93 | 69.21 | 75.87 | 63.63 | |
| N-连接糖基化 (N) | |||||
| Musite-Web[39] | 65.30 | 68.56 | 61.42 | 87.08 | |
| CapsNet[40] | 50.38 | 56.18 | 66.04 | 45.65 | |
| LMNglyPred[43] | 39.34 | 35.17 | |||
| N-linked-PTMGPT2 | 70.92 | 74.21 | 57.49 64.35 | 14.58 87.65 | |
| 精氨酸甲基化 (R) | |||||
| Musite-Web[39] | 67.77 | 68.99 | 85.41 | 57.86 | |
| CapsNet[40] | 65.39 | 65.12 | 90.03 | 51.08 | |
| DeepRMethylSite[44] | 34.02 | 52.49 | 79.89 | 39.08 | |
| PRmePred[45] | 53.76 | 54.33 | 76.45 | 36.22 | |
| CNNArginineMe[46] | 34.55 | 36.98 | 97.42 | 22.82 | |
| RMethyl-PTMGPT2 | 80.51 | 81.32 | 95.14 | 71.01 | |
| 赖氨酸乙酰化 (K) | |||||
| Musite-Web[39] | 21.63 | 36.14 | 94.86 | 22.32 | |
| CapsNet[40] | 21.62 | 36.54 | 93.46 | ||
| GPS-PAIL[47] | 12.35 | 16.14 | 72.63 | 22.71 9.07 | |
| KAcetyl-PTMGPT2 | 22.09 | 40.51 | 96.06 | 25.67 | |
| 泛素化 (K) | |||||
| Musite-Web[39] | 26.24 | 27.67 | 75.67 | 16.93 | |
| DL-Ubiq[48] | 10.91 | 33.37 | |||
| Ubiq-PTMGPT2 | 31.25 | 35.74 | 36.67 80.46 | 23.76 22.97 | |
| O-连接糖基化 (S,T) | |||||
| Musite-Web[39] | 49.89 | 50.29 | 61.64 | 40.38 | |
| OGlyThr[49] | 41.6 | 50.71 | 52.82 | 60.78 | |
| GlyCopp[50] | 41.05 | 14.38 | |||
| O-linked-PTMGPT2 | 58.97 | 61.80 | 50.85 | 46.67 65.14 | |
| S-亚硝基化 (C) | |||||
| PCysMod[51] | 48.53 | 66.67 | 58.79 62.50 | 71.42 | |
| DeepNitro[52] | 79.55 | 45.45 | |||
| PresSNO[53] | 59.45 | 62.50 74.21 | 72.72 | ||
| pIMSNOSite[54] | 69.52 | 84.19 | |||
| SNitro-PTMGPT2 | 55.91 | 17.60 | 14.74 | 21.82 | |
| 丙二酰化 (K) | |||||
| DL-Malosite[55] | 73.61 69.78 | 80.49 77.12 | 84.30 | 77.01 81.94 | |
| Maloy-PTMGPT2 | 73.03 | 78.14 | 72.83 82.95 | 73.85 | |
| 赖氨酸甲基化 (K) | |||||
| Musite-Web[39] | 14.97 | 10.13 | 72.44 | 5.44 | |
| MethylSite[56] | 38.60 | 35.54 | 39.41 | 66.66 | |
| KMethyl-PTMGPT2 | 41.07 | 36.35 | 88.68 | 22.86 | |
| 磷酸化 (S, T) | |||||
| Musite-Web[39] | 17.73 | 12.22 | 6.67 | 72.74 | |
| CapsNet[40] | 6.71 | 71.94 | |||
| PhosphoST-PTMGPT2 | 16.23 16.37 | 12.28 15.35 | 9.14 | 46.97 | |
| Musite-Web[39] | 40.83 | 43.03 | 29.82 | 77.27 | |
| 磷酸化 (Y) | |||||
| CapsNet[40] | 43.85 | 46.61 | 36.58 | 68.18 | |
| PhosphoY-PTMGPT2 | 48.83 | 46.98 | 30.95 | 97.51 | |
| 谷胱甘肽化 | |||||
| DeepGSH[57] | 70.77 | 75.85 | |||
| Glutathio-PTMGPT2 | 74.23 | 75.28 | |||
| ProtTrans-Glutar[58] | 78.28 | 82.37 | 90.07 | 75.89 | |
| 戊二酰化 (K) | |||||
| 62.99 | 59.45 | 68.09 | 79.16 | ||
| Glutary-PTMGPT2 | 69.47 76.00 | 73.78 NR | 71.02 NR | 76.76 81.2 | |
| 酰胺化 | |||||
| Ami-PTMGPT2 | 80.78 | 76.59 | 66.67 | 86.76 | |
| Musite-Web[39] | 35.69 | 47.82 | 79.78 | 49.45 |
Table 2 (continued) | Benchmark dataset results
| PTM | Model MCC | F1Score | Precision | Recall |
| CapsNet40 | 39.81 47.67 | 73.89 | 43.94 | |
| GPS-Palm60 | 24.05 28.49 | 16.69 | 97.19 | |
| Palm-PTMGPT2 | 41.37 | 48.34 | 42.73 55.64 | |
| Proline Hydroxylation (P) | Musite-Web39 | 78.08 | 80.58 98.32 | 67.47 |
| CapsNet40 | 78.87 | 85.92 | 94.66 74.79 | |
| ProHydroxy-PTMGPT2 | 89.89 | 92.30 96.18 | 88.73 | |
| Lysine Hydroxylation (K) | Musite-Web39 | 57.67 63.76 | 76.33 | 72.12 |
| CapsNet40 | 58.87 | 65.92 | 74.66 | |
| LysHydroxy-PTMGPT2 | 66.45 | 68.18 88.23 | 74.79 55.56 | |
| Formylation (K) | CapsNet40 | 40.19 33.33 | 24.09 | 68.34 |
| Musite-Web39 | 39.55 | 28.47 | 23.24 | |
| Formy-PTMGPT2 | 44.58 39.97 | 26.92 | 88.88 77.78 |
Top values for each PTM are represented in bold
表 2 (续表) | 基准数据集结果
| PTM | 模型 MCC | F1分数 | 精确率 | 召回率 |
|---|---|---|---|---|
| CapsNet40 | 39.81 47.67 | 73.89 | 43.94 | |
| GPS-Palm60 | 24.05 28.49 | 16.69 | 97.19 | |
| Palm-PTMGPT2 | 41.37 | 48.34 | 42.73 55.64 | |
| 脯氨酸羟基化 (P) | Musite-Web39 | 78.08 | 80.58 98.32 | 67.47 |
| CapsNet40 | 78.87 | 85.92 | 94.66 74.79 | |
| ProHydroxy-PTMGPT2 | 89.89 | 92.30 96.18 | 88.73 | |
| 赖氨酸羟基化 (K) | Musite-Web39 | 57.67 63.76 | 76.33 | 72.12 |
| CapsNet40 | 58.87 | 65.92 | 74.66 | |
| LysHydroxy-PTMGPT2 | 66.45 | 68.18 88.23 | 74.79 55.56 | |
| 甲酰化 (K) | CapsNet40 | 40.19 33.33 | 24.09 | 68.34 |
| Musite-Web39 | 39.55 | 28.47 | 23.24 | |
| Formy-PTMGPT2 | 44.58 39.97 | 26.92 | 88.88 77.78 |
各PTM类型的最高值以粗体标出
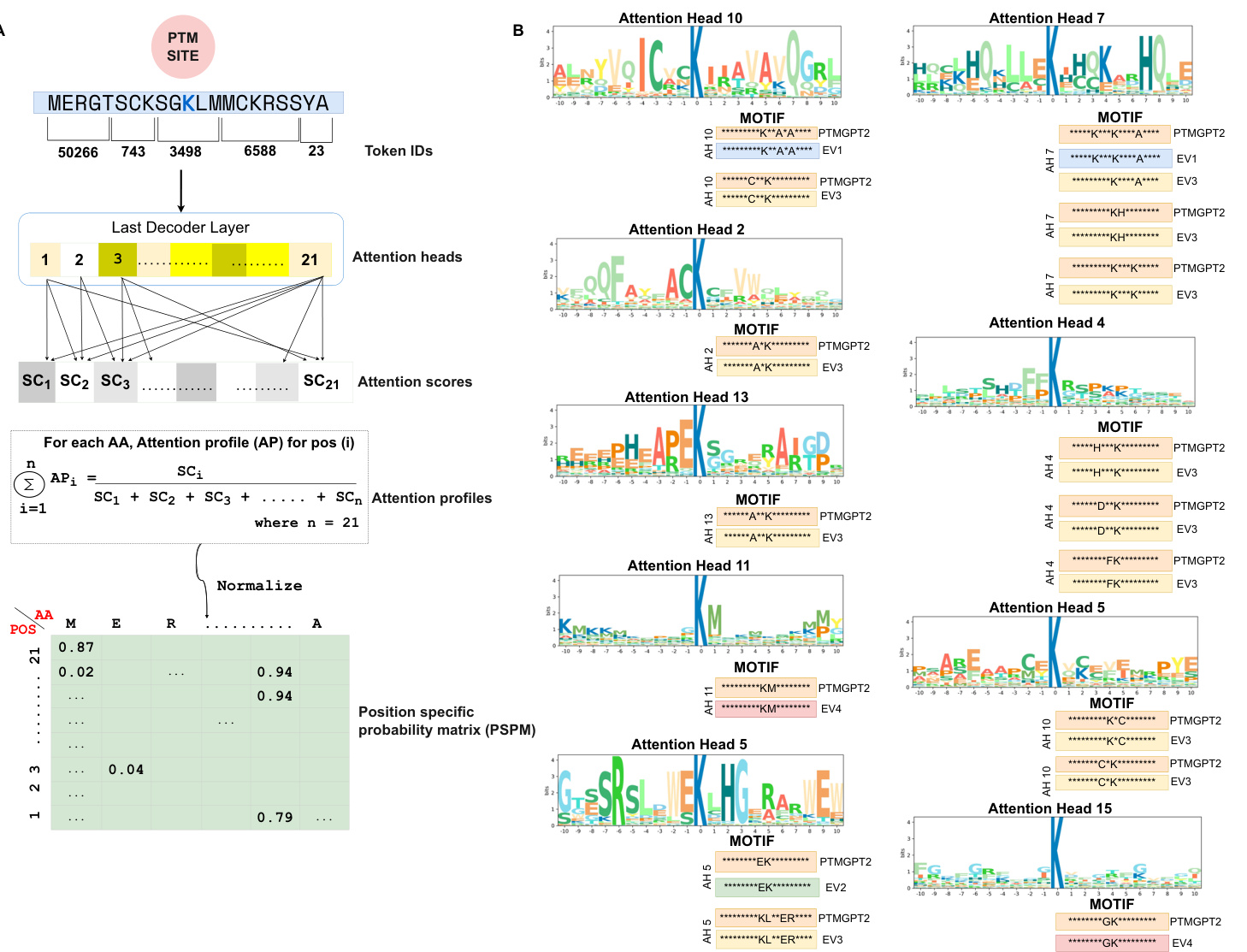
Fig. 2 | Attention head analysis of lysine (K) acetyl ation by PTMGPT2. A Computation of attention scores from the model’s last decoder layer, detailing the process of generating a Position-Specific Probability Matrix (PSPM) for a targeted protein sequence.’SC’ denotes attention scores,’AP’ denotes attention
图 2 | PTMGPT2 对赖氨酸 (K) 乙酰化的注意力头分析。A 从模型最后一个解码器层计算注意力分数,详细说明为目标蛋白质序列生成位置特异性概率矩阵 (PSPM) 的过程。'SC' 表示注意力分数,'AP' 表示注意力概率。
CAMK kinase family, which includes key members like CaMK2 and CAMKL, is crucial in signaling pathways related to neurological disorders, cardiac diseases, and other conditions associated with calcium signaling dys regulation 22. The common motif identified by PTMGPT2 in CAMK was ${\bf R}^{\ast\ast}{\sf S}$ (Arginine at position $^{-2}$ from either a profiles,’AA’ represents an amino acid, and’n’ is the number of amino acids in a sub sequence. B Sequence motifs validated by experimentally verified studies— $\mathrm{EVI}^{61}$ , $\mathrm{EV}2^{62}$ , $\mathrm{EV}3^{63}$ , $\mathrm{EV}4^{47}$ .’AH’ denotes attention head.
CAMK激酶家族, 包括CaMK2和CAMKL等关键成员, 在与神经系统疾病、心脏病及其他钙信号传导失调相关的信号通路中至关重要 22。PTMGPT2在CAMK中识别出的共同基序是 ${\bf R}^{\ast\ast}{\sf S}$ (精氨酸位于 $^{-2}$ 位点)。"AA"代表氨基酸,"n"表示子序列中的氨基酸数量。B 经实验验证研究确认的序列基序—— $\mathrm{EVI}^{61}$ , $\mathrm{EV}2^{62}$ , $\mathrm{EV}3^{63}$ , $\mathrm{EV}4^{47}$ 。"AH"表示注意力头。
phosphor y late d serine or threonine). Further analysis of general protein kinases revealed distinct patterns: DMPK kinase exhibited the motif $\mathbf{RR}^{*}\mathbf{T}$ (Arginine at positions $^{-2}$ and $-3,$ ), MAPKAPK kinase followed the $\mathtt{R}^{*}\mathtt{L S}$ motif (Arginine at position $^{-3}$ and leucine at position −1), AKT kinase was characterized by the ${\tt R}^{*}{\tt R S}$ motif (Arginine at positions $^{-1}$ and $^{-3}$ ), CK1 kinase showed $\mathsf{K}^{*}\mathsf{K}^{**}\mathsf{S}/\mathsf{T}$ (Lysine at positions $^{-3}$ and $-5.$ ), and CK2 kinase was defined by the $\mathsf{S D}^{*}\mathbf{E}$ motif (Aspartate at position $^{+1}$ and glutamate at position $^{+3}$ ). These comparisons underscored PTMGPT2’s ability to accurately identify motifs associated with diverse kinase groups and PTM types. PSPM matrices, corresponding to 20 attention heads across all 19 PTM types, are detailed in Supplementary Data 1. These insights are crucial for deciphering the intricate mechanisms underlying protein modifications. Consequently, this analysis, driven by the PTMGPT2 model, forms a core component of our exploration into the contextual relationships between protein sequences and their predictive labels.
磷酸化的丝氨酸或苏氨酸)。对通用蛋白激酶的进一步分析揭示了不同的模式:DMPK激酶呈现基序 $\mathbf{RR}^{*}\mathbf{T}$(精氨酸位于 $^{-2}$ 和 $-3$ 位),MAPKAPK激酶遵循 $\mathtt{R}^{*}\mathtt{L S}$ 基序(精氨酸位于 $^{-3}$ 位,亮氨酸位于-1位),AKT激酶以 ${\tt R}^{*}{\tt R S}$ 基序为特征(精氨酸位于 $^{-1}$ 和 $^{-3}$ 位),CK1激酶显示 $\mathsf{K}^{*}\mathsf{K}^{**}\mathsf{S}/\mathsf{T}$(赖氨酸位于 $^{-3}$ 和 $-5$ 位),CK2激酶则由 $\mathsf{S D}^{*}\mathbf{E}$ 基序定义(天冬氨酸位于 $^{+1}$ 位,谷氨酸位于 $^{+3}$ 位)。这些比较结果突显了PTMGPT2准确识别不同激酶组和PTM类型相关基序的能力。对应所有19种PTM类型中20个注意力头的PSPM矩阵详见补充数据1。这些发现对解析蛋白质修饰的复杂机制至关重要。因此,这项由PTMGPT2模型驱动的分析构成了我们探索蛋白质序列与其预测标签间上下文关系的核心组成部分。
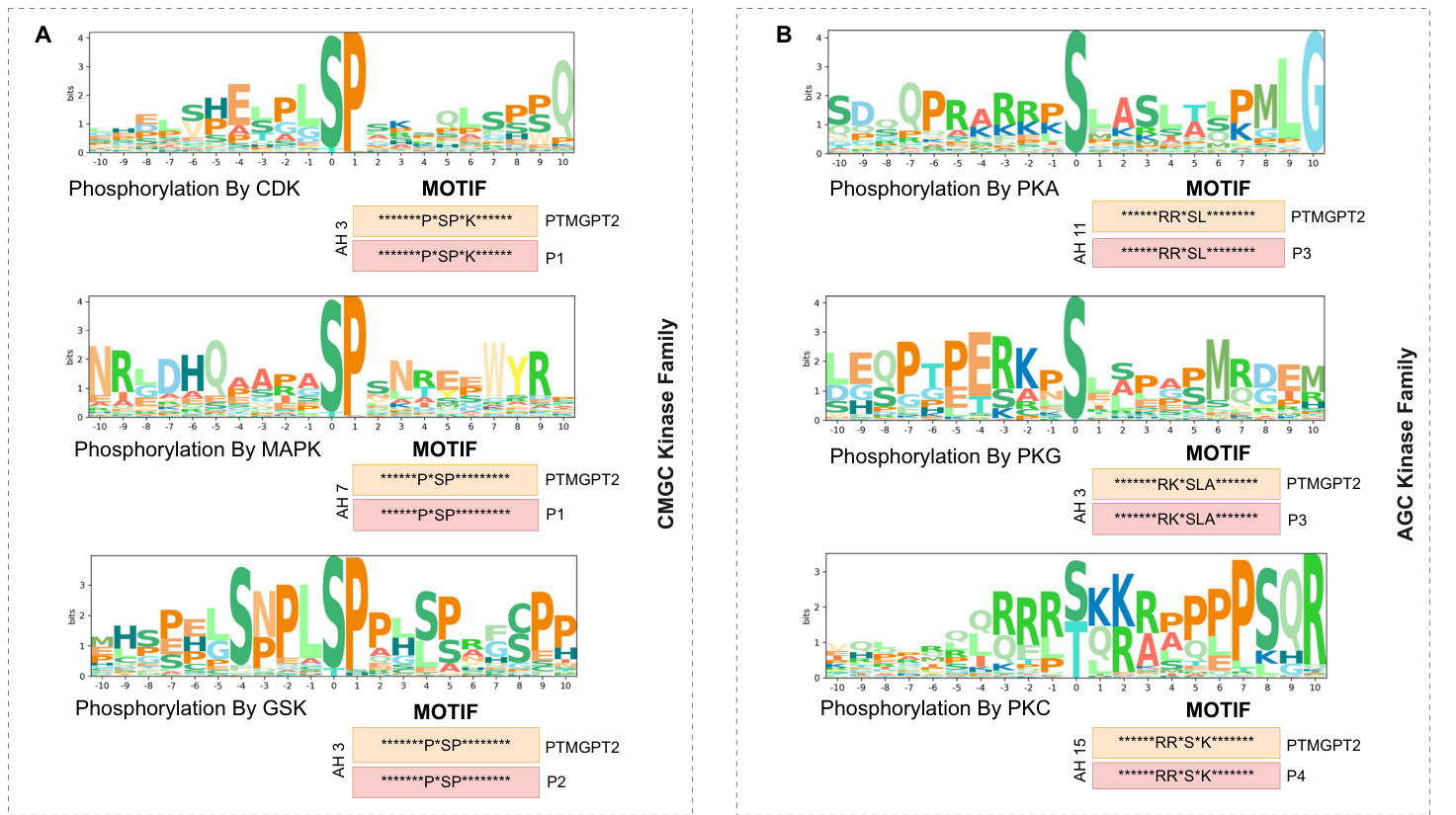
Fig. 3 | Attention head analysis of the CMGC kinase family and the AGC kinase family by PTMGPT2. A Motifs from CMGC kinase family validated against ${\mathsf{P}}1^{64}$ and ${\mathsf{P}}2^{65}$ . B AGC kinase family motifs validated against $\mathsf{P}3^{66}$ and $\mathbf{P}4^{67}$ . The’P*SP’ motif is
图 3 | PTMGPT2对CMGC激酶家族和AGC激酶家族的注意力头分析。A 基于 ${\mathsf{P}}1^{64}$ 和 ${\mathsf{P}}2^{65}$ 验证的CMGC激酶家族基序。B 基于 $\mathsf{P}3^{66}$ 和 $\mathbf{P}4^{67}$ 验证的AGC激酶家族基序。"P*SP"基序是
Recent uniprot entries validate PTMGPT2’s robust generalization abilities
最新UniProt条目验证PTMGPT2的稳健泛化能力
To demonstrate PTMGPT2’s robust predictive capabilities on unseen datasets, we extracted proteins recently released on UniProt, strictly selecting those added after June 1, 2023, to validate the model’s performance. We ensured these proteins were not present in the training or benchmark datasets from DBPTM (version May 2023), which was a crucial step in the validation process. A total of 31 proteins that met our criteria were identified, associated with PTMs such as phosphor yl ation (S, T, Y), methyl ation (K), and acetyl ation (K). The accurate prediction of PTMs in recently identified proteins not only validates the effectiveness of our model but also underscores its potential to advance research in protein biology and PTM site identification. These predictions are pivotal for pinpointing the precise locations and characteristics of modifications within the protein sequences, which are crucial for verifying PTMGPT2’s performance. The predictions for all 31 proteins, along with the ground truth, are detailed in Supplementary Table S1−S5.
为验证PTMGPT2在未知数据集上的强大预测能力,我们从UniProt中提取了近期发布的蛋白质,严格筛选2023年6月1日后新增的蛋白质以评估模型性能。我们确保这些蛋白质未出现在DBPTM(2023年5月版)的训练集或基准数据集中,这是验证过程中的关键步骤。最终确定了31个符合标准的蛋白质,涉及磷酸化(S, T, Y)、甲基化(K)和乙酰化(K)等翻译后修饰。对新发现蛋白质中PTM的准确预测不仅验证了我们模型的有效性,更凸显了其在推动蛋白质生物学研究和PTM位点识别方面的潜力。这些预测对于精确定位蛋白质序列中修饰的具体位置和特征至关重要,为验证PTMGPT2的性能提供了关键依据。所有31个蛋白质的预测结果及真实数据详见补充表S1-S5。
PTMGPT2 identifies mutation hotspots in ph os ph o sites of TP53, BRAF, and RAF1 genes
PTMGPT2 识别 TP53、BRAF 和 RAF1 基因磷酸化位点的突变热点
Protein PTMs play a vital role in regulating protein function. A key aspect of PTMs is their interplay with mutations, particularly near common in the CMGC kinase family, whereas the’R**S’ motif is common in the AGC kinase family.’AH’ denotes attention head.
蛋白质翻译后修饰 (PTMs) 在调控蛋白质功能中起着至关重要的作用。PTMs 的一个关键方面是其与突变的相互作用,特别是在 CMGC 激酶家族中常见的 (R**S) 基序,而 (R**S) 基序在 AGC 激酶家族中常见。(AH) 表示注意力头。
modification sites, where mutations can significantly impact protein function and potentially lead to disease. Previous studies23–25 indicate a strong correlation between pathogenic mutations and proximity to ph os ph os erin e sites, with over $70%$ of PTM-related mutations occurring in phosphor yl ation regions. Therefore, our study primarily targets ph os ph os erin e sites to provide a more in-depth understanding of PTM-related mutations. This study aims to evaluate PTMGPT2’s ability to identify mutations within 1−8 residues flanking a ph os ph os erin e site, without explicit mutation site annotations during training. For this, we utilized the dbSNP database26, which includes information on human single nucleotide variations linked to both common and clinical mutations. $T P53^{27}$ is a critical tumor suppressor gene, with mutations in TP53 being among the most prevalent in human cancers. When mutated, TP53 may lose its tumor-suppressing function, leading to uncontrolled cell proliferation. $B R A F^{28}$ is involved in intracellular signaling critical for cell growth and division. BRAF mutations, especially the V600E mutation, are associated with various cancers such as melanoma, thyroid cancer, and colorectal cancer. $R A F\boldsymbol{1}^{29}$ plays a role in the RAS/MAPK signaling pathway. While RAF1 mutations are less common in cancers compared to BRAF, abnormalities in RAF1 can contribute to oncogene s is and genetic disorders like Noonan syndrome, characterized by developmental abnormalities.
修饰位点, 即突变可能显著影响蛋白质功能并潜在导致疾病的位置。先前研究[23-25] 表明致病突变与磷酸化位点邻近性存在强相关性, 超过 $70%$ 的翻译后修饰相关突变发生在磷酸化区域。因此, 我们的研究主要聚焦于磷酸化位点, 以提供对翻译后修饰相关突变更深入的理解。本研究旨在评估 PTMGPT2 在训练期间未获得明确突变位点标注的情况下, 识别磷酸化位点侧翼 1-8 个残基内突变的能力。为此, 我们利用 dbSNP 数据库 [26] , 其中包含与常见突变及临床突变相关的人类单核苷酸变异信息。 $TP53^{27}$ 是关键肿瘤抑制基因, TP53 突变是人类癌症中最普遍的突变类型之一。当 TP53 发生突变时, 可能丧失抑癌功能, 导致细胞增殖失控。 $BRAF^{28}$ 参与调控细胞生长和分裂至关重要的细胞内信号传导。BRAF 突变 (尤其是 V600E 突变) 与黑色素瘤、甲状腺癌和结直肠癌等多种癌症相关。 $RAF1^{29}$ 在 RAS/MAPK 信号通路中发挥作用。虽然 RAF1 突变在癌症中较 BRAF 突变少见, 但 RAF1 异常可能导致肿瘤发生和努南综合征等以发育异常为特征的遗传性疾病。
PTMGPT2’s analysis of the TP53 gene revealed a complex pattern of ph os ph o site mutations depicted in Fig. 5A, including G374, K370, and H368, across multiple cancer types25. This is validated by dbSNP data, indicating that 21 of the top 28 mutations with the highest number of adjacent modifications occur in the tumor suppressor protein TP53. The RAF1 gene, a serine/threonine kinase, exhibits numerous mutations, many of which are associated with disrupted MAPK activity due to altered recognition and regulation of PTMs. In our analysis of RAF1 S259 phosphor yl ation, PTMGPT2 precisely identified mutations directly on S259 and in adjacent hotspots at residues S257, T258, and P261 depicted in Fig. 5B. These findings are consistent with genetic studies29,30 linking RAF1 mutations near S259 to Noonan and LEOPARD Syndrome. Furthermore, in BRAF, another serine/ threonine kinase, PTMGPT2’s analysis of the S602 phosphor yl ation site revealed mutations in flanking residues (1–7 positions) such as D594N, L597Q, V600E, V600G, and $\mathsf{K}60\mathsf{1}\mathsf{E}^{23}$ shown in Fig. 5C. These mutations, particularly those activating BRAF functions, are found in over $60%$ of melanomas 28. Heatmap plots and line plots for remaining genes in dbSNP, and a bar chart depicting the selected genes for analysis, are provided in Supplementary Figs. S1−S19. These results demonstrate PTMGPT2’s proficiency not only in predicting PTM sites but also in identifying potential mutation hotspots around these sites.
PTMGPT2对TP53基因的分析揭示了复杂的磷酸化位点突变模式,如图 5A 所示,包括跨多种癌症类型的 G374、K370 和 H368 位点突变 [25]。该结果通过 dbSNP 数据得到验证,表明在相邻修饰数量最多的 28 个突变中,有 21 个发生在肿瘤抑制蛋白 TP53 上。丝氨酸/苏氨酸激酶 RAF1 基因存在大量突变,其中许多突变因蛋白质翻译后修饰 (PTM) 的识别与调控改变而导致 MAPK 活性紊乱。在对 RAF1 S259 磷酸化位点的分析中,PTMGPT2 精准识别了 S259 直接位点突变及相邻残基 S257、T258 和 P261 的热点突变(图 5B)。这些发现与遗传学研究 [29,30] 一致,证实 RAF1 S259 附近突变与努南综合征和豹皮综合征相关。此外在另一种丝氨酸/苏氨酸激酶 BRAF 中,PTMGPT2 对 S602 磷酸化位点的分析揭示了侧翼残基(1-7 位点)突变,包括 D594N、L597Q、V600E、V600G 和 $\mathsf{K}60\mathsf{1}\mathsf{E}^{23}$(图 5C)。这些突变(尤其是激活 BRAF 功能的突变)在超过 $60%$ 的黑色素瘤中被发现 [28]。dbSNP 中其余基因的热图与折线图,以及分析基因筛选的条形图详见补充图 S1-S19。这些结果证明 PTMGPT2 不仅能预测蛋白质翻译后修饰位点,还能有效识别这些位点周围的潜在突变热点。

ig. 4 | Attention head analysis of the CAMK kinase family and general protein kinases by PTMGPT2. A CAMK kinase family motifs validated against $\mathsf{P}5^{68}$ . B General protein kinase motifs validated against $\mathsf{P}6^{69}$ , $\mathbf{P}7^{70}$ , and $\mathbf{P}\mathbf{8}^{71}$ . The’ $\mathbf{R}^{**}\mathbf{S}^{\prime}$ motif is common in the CAMK kinase family. ’AH’ denotes attention head.
图 4 | PTMGPT2对CAMK激酶家族及通用蛋白激酶的注意力头分析。(A) 基于 $\mathsf{P}5^{68}$ 验证的CAMK激酶家族基序。(B) 基于 $\mathsf{P}6^{69}$ 、 $\mathbf{P}7^{70}$ 和 $\mathbf{P}\mathbf{8}^{71}$ 验证的通用蛋白激酶基序。' $\mathbf{R}^{**}\mathbf{S}^{\prime}$ '基序在CAMK激酶家族中普遍存在。'AH'表示注意力头。
Discussion
讨论
GPT models have significantly advanced the state of NLP by demonstrating the power of the transformer architecture, the efficacy of pretraining on a large corpus of data, and the versatility of language models across a range of tasks through transfer learning. In this paper, we proposed PTMGPT2 for protein PTM site prediction in a way that reformulates protein classification tasks as protein label generation. We discovered that PTMGPT2, when subjected to prompt-based finetuning on large-scale datasets and tested on external benchmarks, demonstrates notable improvement in prediction accuracy, with an average improvement of $5.45%$ in MCC, underscoring the efficacy of protein language as a robust yet powerful descriptor for PTM site prediction. The crucial role of structuring an informative prompt, which accurately captured the dependencies between protein sequence and its corresponding label, played a significant role in building an accurate generative model. Along with generating correct sequence labels for each PTM type, PTMGPT2 was able to precisely interpret attention scores as motifs and analyze the significance of having a pathogenic mutation directly on the site of modification or within a possible recognition area of the site of modification. This ability aids in exploring internal data distribution related to biochemical significance.
GPT模型通过展现Transformer架构的强大能力、在大规模数据语料上进行预训练的有效性,以及通过迁移学习在一系列任务中语言模型的多功能性,显著推进了自然语言处理(NLP)的发展。在本文中,我们提出了PTMGPT2用于蛋白质翻译后修饰(PTM)位点预测,其方式是将蛋白质分类任务重新定义为蛋白质标签生成。我们发现,当PTMGPT2在大规模数据集上基于提示进行微调并在外部基准测试中进行评估时,预测准确性显示出显著提升,MCC平均提高$5.45%$,这强调了蛋白质语言作为PTM位点预测的强大描述符的有效性。构建信息丰富的提示的关键作用,准确捕捉了蛋白质序列与其相应标签之间的依赖关系,在构建准确的生成模型中发挥了重要作用。除了为每种PTM类型生成正确的序列标签外,PTMGPT2还能够精确将注意力分数解释为基序,并分析在修饰位点直接或可能在修饰位点的识别区域内发生致病突变的重要性。这种能力有助于探索与生物化学意义相关的内部数据分布。
We employed PTMGPT2 for 19 different PTM types, including phosphor yl ation, N-linked g lycos yl ation, N6-acetyl lys in e, methylarginine, succ in yl ation, sumo yl ation, lysine acetyl ation, ubiquitination, O-linked g lycos yl ation, S-nitro syl ation, mal on yl ation, methyllysine, glut at hi on yl ation, glut ary la tion, amidation, S-palm i to yl ation, hydroxyl ation, and for myl ation. The comparative results demonstrate that PTMGPT2 outperforms existing deep-learning methods and tools in most cases, offering promising prospects for practical applications. PTMGPT2s performance, categorized by species and evaluated using MCC, F1 Score, Precision, and Recall for all 19 PTMs, is listed in Supplementary Tables S6−S23. One limitation of our approach is the constrained exploration of prompt designs for certain PTM types, particularly in instances where PTMGPT2 did not surpass the performance of competing methods. We plan to investigate additional prompts and model tuning specifically designed for PTMs such as phosphor yl ation (S,T) in future versions of our work. Another limitation associated with PTM sites is the presence of false negatives in the training data. To mitigate the impact of false negatives, we plan in our future work to incorporate methods similar to outlier detection and anomaly detection, which focus on modeling the distribution of positive data, instances where PTMs are known to occur. By not relying on negative data, which can be sparsely labeled or mis classified in biological datasets, it is possible to avoid the common pitfalls of training models with false negatives. The reason behind implementing task-specific tuning for each PTM type, rather than utilizing shared weights among these PTMs, was that a single model using shared weights would require specially designed prompts to accurately handle multiple concurrent PTM types on the same residue. Moreover, in our motif identification analysis, utilizing a shared-weight model could complicate the interpretation of which residues influenced specific types of modifications. PTMGPT2 marks a major leap in protein PTM site prediction, laying the groundwork for future studies on protein sequence and function through the analysis of complex GPT-based attention mechanisms and their real-world applications. The autoregressive nature of PTMGPT2, which predicts the next token based on all previously observed tokens in a prompt, is particularly suited for tasks involving sequential data like protein sequences. Regarding the potential application of other large language models, it is plausible that similar enhancements could be achieved if these models are properly trained and fine-tuned for specific tasks in bioinformatics. Future efforts will focus on exploring and restructuring prompts, a strategy aimed at improving the accuracy of PTM site predictions within the protein domain.
我们采用PTMGPT2处理19种不同类型的翻译后修饰 (PTM) ,包括磷酸化、N-连接糖基化、N6-乙酰赖氨酸、甲基精氨酸、琥珀酰化、苏素化、赖氨酸乙酰化、泛素化、O-连接糖基化、S-亚硝基化、丙二酰化、甲基赖氨酸、谷胱甘肽化、戊二酰化、酰胺化、S-棕榈酰化、羟基化和甲酰化。对比结果表明,PTMGPT2在多数情况下优于现有深度学习方法和工具,为实际应用提供了良好前景。PTMGPT2按物种分类的性能,以及使用MCC、F1分数、精确度和召回率对所有19种PTM的评估结果,列于补充表S6-S23。我们方法的一个局限性在于对某些PTM类型的提示设计探索有限,特别是在PTMGPT2未超越竞争方法性能的情况下。我们计划在后续版本中专门针对磷酸化 (S,T) 等PTM研究更多提示设计和模型调优。另一个与PTM位点相关的局限是训练数据中存在假阴性。为减轻假阴性的影响,我们计划在未来工作中采用类似离群值检测和异常检测的方法,这些方法侧重于对阳性数据(即已知发生PTM的实例)的分布建模。通过不依赖生物数据集中可能标注稀疏或分类错误的阴性数据,可以避免使用假阴性训练模型的常见缺陷。对每种PTM类型实施任务特异性调优而非使用共享权重的原因在于:使用共享权重的单一模型需要专门设计的提示才能准确处理同一残基上的多种并发PTM类型。此外,在基序识别分析中,使用共享权重模型可能使特定修饰类型受影响残基的解读复杂化。PTMGPT2标志着蛋白质PTM位点预测的重大飞跃,通过分析基于GPT的复杂注意力机制及其实际应用,为未来研究蛋白质序列和功能奠定基础。PTMGPT2的自回归特性(根据提示中所有已观测token预测下一个token)特别适用于蛋白质序列等序列数据任务。关于其他大语言模型的潜在应用,若这些模型经过适当训练并针对生物信息学特定任务进行微调,实现类似增强效果是可行的。未来工作将重点探索和重构提示策略,旨在提升蛋白质领域PTM位点预测的准确性。

Fig. 5 | PTMGPT2 analysis of mutation distribution around PTM sites. Heatmaps and corresponding line plots illustrate the probability and impact of mutations within the recognition sites of ph os ph os erin es across TP53, RAF1, and BRAF genes. The heatmaps’ X-axes display the wild-type sequence while the Y-axes represent the 20 standard amino acids; yellow indicates the presence of mutations and darker shades indicate their absence. A For TP53 S371 ph os ph os erin e, PTMGPT2 predicts mutations predominantly in the 1−2 flanking residues. The line plot shows the average effect of these mutations by position, and the sequence plot reveals
图 5 | PTMGPT2对翻译后修饰位点周围突变分布的分析。热图及对应折线图展示了TP53、RAF1和BRAF基因中磷酸化识别位点内突变的概率与影响。热图X轴显示野生型序列,Y轴代表20种标准氨基酸;黄色表示存在突变,颜色越深表示突变缺失。A 针对TP53 S371磷酸化位点,PTMGPT2预测突变主要分布在1-2个侧翼残基。折线图按位置显示这些突变的平均效应,序列图揭示
predicted mutation hotspots directly on S371 and 1−2 residues from S371 across multiple human diseases. B Analysis of RAF1 S259 ph os ph os erin e, showing a concentrated mutation effect at the S259 and its immediate vicinity. C For BRAF S602 ph os ph os erin e, PTMGPT2 identifies a broader distribution of mutations within the 1−7 flanking residues, with the line plot indicating significant mutation impacts at positions close to the S602. In the sequence plots,’M’ represents a mutation, and’PS’ indicates a phosphor y late d serine residue. Source data are provided as a Source Data file.
在多个人类疾病中,S371及其1-2个残基范围内的预测突变热点。B RAF1 S259磷酸化位点分析显示,S259及其紧邻区域存在集中突变效应。C 对于BRAF S602磷酸化位点,PTMGPT2识别出1-7个侧翼残基内更广泛的突变分布,折线图显示靠近S602的位点具有显著突变影响。序列图中"M"代表突变,"PS"表示磷酸化丝氨酸残基。源数据文件详见Source Data。
Methods
方法
Prompt design
提示设计
In designing the prompt, our aim was to enable PTMGPT2 to distinguish between modified and unmodified protein sequences and design a flexible approach that can generalize to new classification tasks without the need for task-specific classification heads. The key was selecting tokens that accurately represented the protein sub sequence and capture its contextual dependency with generated tokens. We incorporated two special tokens:
在提示设计过程中,我们的目标是使PTMGPT2能够区分修饰与未修饰的蛋白质序列,并设计一种可推广至新分类任务的灵活方法,无需依赖特定任务的分类头。关键在于选择能准确表征蛋白质子序列并捕获其与生成token间上下文依赖关系的token。我们引入了两个特殊token:<start of text> 和 <endoftext \$>\$ 分别标记提示的起始与结束。同时整合了四个自定义token:'SEQUENCE:' 表示蛋白质子序列,'LABEL:' 表示PTM真实标签,'POSITIVE' 代表修饰序列,'NEGATIVE' 代表未修饰序列。提示1包含四个自定义token flanking 21个氨基酸长度的蛋白质子序列。提示2将21长度子序列排列为两个10聚体,标定修饰或未修饰的中心残基。与之对比,提示3仅使用特殊token,省略了自定义token,但保持相同的21氨基酸子序列。提示4仅包含特殊token,但将序列扩展至51个氨基酸。最后,提示5将特殊token与自定义token结合,使用51长度蛋白质序列。所有用于微调PTMGPT2的提示展示于图1A。推理提示展示于补充图S20。
Dataset preparation
数据集准备
Our dataset was compiled from DBPTM, a resource offering both experimentally verified training and non-homologous benchmark datasets. We extracted information regarding the UniProt ID, modified residue, and modification type for 19 PTM types from DBPTM, which were then used to retrieve full-length protein sequences from Uniprot, available at https://www.uniprot.org/id-mapping. This step was necessary, as DBPTM does not provide full-length sequences. The training and benchmark datasets were compiled based on the categori zat ions pre-established by DBPTM. These specific PTMs were selected due to their high data volume, aligning with the requirement of GPT-based models for substantial data to develop an effective predictive model. Positive samples were prepared by extracting 21- length sub sequences centered on modified residues. Negative samples were prepared by extracting 21-length sub sequences from unmodified residues (amino acid positions not annotated as modified) for each distinct PTM type, using an approach identical to that used for positive samples. After combining both positive and negative sub sequences for each PTM, we compiled them into a fasta file. To reduce sequence redundancy, sequences with over $30%$ similarity were removed using CD-hit31 for each PTM as shown in Fig. 1B. This process resulted in highly imbalanced yet refined training datasets for 19 PTMGPT2 models. Performance results for PTMGPT2 with CD-hit similarity cutoffs of $40%$ and $50%$ have been included in Supplementary Table S24 to provide a detailed analysis of how different thresholds affect the model’s performance. To benchmark against models that require specific input formats, our data preparation had to align with their unique requirements, whether they needed variable-length sequences or symbol-based inputs. UniProt identifiers were mapped to acquire full-length sequences, from which method-specific sub sequences were generated. Subsequently, we excluded any protein sub sequences in the benchmark dataset that overlapped with our training dataset. Detailed data statistics for each PTM type are provided in Supplementary Tables S25 and S26. Data preparation pipelines for both training and benchmark datasets are shown in Supplementary Figs. S21 and S22.
我们的数据集编译自DBPTM,该资源提供经实验验证的训练集和非同源基准数据集。我们从DBPTM中提取了19种蛋白质翻译后修饰(PTM)类型的UniProt ID、修饰残基和修饰类型信息,随后通过https://www.uniprot.org/id-mapping从Uniprot获取全长蛋白质序列。这一步骤是必要的,因为DBPTM不提供全长序列。训练集和基准数据集的编制基于DBPTM预设的分类体系。选择这些特定PTM类型是因为其数据量充足,符合基于GPT的模型需要大量数据来开发有效预测模型的要求。
正样本通过提取以修饰残基为中心的21长度亚序列制备。负样本则采用与正样本相同的方法,从每种PTM类型的未修饰残基(未标注为修饰的氨基酸位点)中提取21长度亚序列。将每种PTM的正负亚序列合并后,我们将其编译为fasta文件。为降低序列冗余度,使用CD-hit31移除了相似度超过 $30%$ 的序列(如图1B所示)。该过程最终为19个PTMGPT2模型生成了高度不平衡但精炼的训练数据集。PTMGPT2在CD-hit相似度阈值为 $40%$ 和 $50%$ 时的性能结果已列入附表S24,以详细分析不同阈值对模型性能的影响。
为对需要特定输入格式的模型进行基准测试,我们的数据制备必须符合其独特要求——无论是需要可变长度序列还是基于符号的输入。通过映射UniProt标识符获取全长序列,并据此生成方法特异性亚序列。随后,我们排除了基准数据集中与训练数据集存在重叠的任何蛋白质亚序列。各PTM类型的详细数据统计信息见附表S25和S26。训练集和基准数据集的数据制备流程分别见附图S21和S22。
Vocabulary encoding
词汇编码
We utilized the pre-trained tokenizer, fine-tuned on the BPE sub-word token iz ation algorithm, to encode protein sequences. Unlike traditional methods that assign a unique identifier to each residue, this approach recognizes entire motifs as unique identifiers during tokenization. This not only retains evolutionary information but also highlights conserved motifs within sequences. As a result, this technique offers advantages over one-hot token iz ation and alleviates outof-vocabulary issues. On average, a token is represented by a motif of four amino acids. We expanded the pre-trained tokenizer’s vocabulary to incorporate four custom tokens for prompt fine-tuning: ‘POSITIVE’, ‘NEGATIVE’, ‘SEQUENCE:’, and ‘LABEL:’, in addition to two special tokens: ‘<start of text $>^{\prime}$ and ‘<endoftext $>^{\prime}$ . These tokens play a crucial role in constructing the prompts for output generation, where ‘POSITIVE’ and ‘NEGATIVE’ are the tokens generated by PTMGPT2, represented by token IDs 50262 and 50263, respectively. Furthermore, ‘SEQUENCE:’ and’LABEL:’ are instrumental in the finetuning process, represented by token IDs 50260 and 50261, respectively. This process resulted in a comprehensive vocabulary for PTMGPT2, with a total of 50,264 tokens.
我们使用基于BPE子词token化算法微调的预训练分词器对蛋白质序列进行编码。与传统方法为每个残基分配唯一标识符不同,该方法在token化过程中将完整基序识别为唯一标识符。这不仅保留了进化信息,还突出了序列中的保守基序。因此,该技术相比独热编码具有优势,并缓解了未登录词问题。平均每个token对应由四个氨基酸组成的基序。我们扩展了预训练分词器的词汇表,除两个特殊token '
Model training and inference
模型训练与推理
We began by instant i a ting the PROTGPT2 model and tokenizer, sourced from Hugging Face 32. We initialized the model with pre-trained weights and fine-tuned it for our label generation task using an unsupervised approach, retaining tokens during training to learn sequencelabel dependency. During inference, the ‘POSITIVE’, ‘NEGATIVE’, and ‘<endoftext $>^{\prime}$ tokens were excluded, allowing the model to generate labels that identify whether a protein sequence is modified or unmodified as shown in Fig. 1C. We utilized Hugging Face’s trainer object to establish the training loop. Training each PTM model lasted 200 epochs, using a batch size of 128 per device, a weight decay of 0.01 for the Adam optimizer, and a learning rate of 1e-03, consistent with the original pre-trained model. Negative log-likelihood was utilized as the loss function for fine-tuning and checkpoints were saved every 500 steps. The primary objective during inference was to remove the’POSITIVE’ and’NEGATIVE’ tokens and utilize greedy sampling to generate the most probable token associated to a modification, which was then compared with the ground truth. To evaluate the performance of PTMGPT2, we selected the best performing checkpoint on the basis of MCC, F1Score, precision, and recall on external benchmark dataset for each PTM type. All PTMGPT2 models were trained and bench-marked using NVIDIA A100 80GB and NVIDIA RTX A6000 48GB GPUs.
我们首先从Hugging Face [32] 加载了PROTGPT2模型和分词器 (tokenizer) 。使用预训练权重初始化模型,并通过无监督方法针对标签生成任务进行微调,在训练过程中保留token以学习序列标签依赖关系。推理阶段排除'POSITIVE'、'NEGATIVE'和'
Benchmark comparison
基准测试对比
For CapsNet, developed in 2018, we retrained models for sumo yl ation, N-linked g lycos yl ation, methyl ation, acetyl ation, phosphor yl ation, and S-palm i to yl ation. Similarly, for MusiteDeep, introduced in 2020, the models were retrained for sumo yl ation, N-linked g lycos yl ation, methyl ation, acetyl ation, phosphor yl ation, S-palm i to yl ation, ubiquitination, and O-linked g lycos yl ation. The rationale for retraining these models included the availability of their training code and their documentation of multiple PTMs, which facilitated the comparison of varied PTMs. Additionally, the presence of overlapping protein sequences between our benchmark data and their training data necessitated the removal of overlapping data points and the retraining of their models from scratch using unique training sequences. This approach ensured a clearer demonstration of each method’s intrinsic advantages without the influence of data overlap. To address the potential implications of outdated data, although the original publications for CapsNet and MusiteDeep did not include models for hydroxyl ation and for myl ation, we trained models from scratch using updated training data from DBPTM for these PTMs. This allowed for comparison with PTMGPT2 and was also required by the absence of other functional existing methods for these modifications. Methods introduced from 2021 onwards were not retrained, as they utilized more recent protein sequence data; therefore, they were compared using our common benchmark data. For other methods published before 2021, whose training code was not publicly available, we resorted to using their online webservers for comparison. Bar charts for benchmark comparison are provided in Supplementary Figs. S23−S41.
对于2018年开发的CapsNet, 我们针对sumo化、N-连接糖基化、甲基化、乙酰化、磷酸化和S-棕榈酰化重新训练了模型。同样地, 对于2020年推出的MusiteDeep, 我们针对sumo化、N-连接糖基化、甲基化、乙酰化、磷酸化、S-棕榈酰化、泛素化和O-连接糖基化重新训练了模型。重新训练这些模型的理由包括: 可获得其训练代码、它们对多种PTM的记录, 这有助于比较不同的PTM。此外, 由于我们的基准数据与其训练数据中存在重叠的蛋白质序列, 需要去除重叠的数据点, 并使用独特的训练序列从头开始重新训练它们的模型。这种方法确保了在没有数据重叠影响的情况下, 更清晰地展示每种方法的内在优势。为了解决数据过时的潜在影响, 尽管CapsNet和MusiteDeep的原始出版物未包含羟基化和豆蔻酰化的模型, 但我们使用来自DBPTM的更新训练数据为这些PTM从头开始训练了模型。这使得能够与PTMGPT2进行比较, 并且由于缺乏其他针对这些修饰的功能性现有方法, 这也是必要的。2021年之后推出的方法未进行重新训练, 因为它们使用了更新的蛋白质序列数据; 因此, 使用我们的共同基准数据进行比较。对于2021年之前发布的其他方法, 由于训练代码未公开, 我们使用其在线网络服务器进行比较。基准比较的条形图见补充图S23−S41。
PSPM and attention head visualization
PSPM 与注意力头可视化
We proceeded by preparing the input sequence, prefixed and suffixed with custom tokens that delineated the sequence and label within the prompt. This formatted text was tokenized into a tensor of token IDs, which the model processed to produce attention scores. Since tokens represent motifs and the self-attention scores in GPT2 decoders operate between tokens, we calculate the attention scores for individual amino acids. To achieve this, we d is aggregate the attention assigned to each token back down to the constituent amino acids. This d is aggregation is performed such that each amino acid within a token is assigned the same attention score as the token’s attention score. The process of transforming attention scores into attention profiles began with the initialization of a matrix to collate attention scores across the 20 standard amino acids for 21 sequence positions. The attention scores, derived from the previous step, were iterative ly accumulated for each amino acid at each position in the sequence, skipping any instances of the amino acid’X’, typically indicative of an unknown amino acid. This resulted in a matrix reflecting the weighted significance of each amino acid at every position. Normalization of this matrix against the sum of attention scores per position yielded a relative attention profile. Next step was to transform sequences and their corresponding attention profiles into a PSPM Matrix. Initially, this involved accumulating attention profiles for each amino acid at their specific positions in the sequences. This was achieved by iterating over the sequences, ensuring that the length of each sequence matches its associated attention profiles to maintain data integrity. The Logomaker Python package33 was used to generate protein sequence logos. PSPM was provided as input to the Logomaker object. The algorithm for generating a PSPM is detailed in Supplementary Fig. S42.
我们首先准备输入序列,在提示文本中用自定义token作为前缀和后缀来划分序列和标签。这段格式化文本被转换为token ID张量,模型通过处理这些张量生成注意力分数。由于token代表基序,且GPT2解码器中的自注意力机制作用于token之间,因此我们需要计算单个氨基酸的注意力分数。为实现这一目标,我们将分配给每个token的注意力分数分解至其组成的氨基酸。这种分解方式使得同一token内的所有氨基酸都被赋予与该token相同的注意力分数。
将注意力分数转换为注意力谱的过程始于初始化一个矩阵,该矩阵用于汇总21个序列位置上20种标准氨基酸的注意力分数。基于前一步骤得到的注意力分数,我们对序列中每个位置的每个氨基酸进行迭代累加(跳过代表未知氨基酸的"X"),最终形成反映每个位置氨基酸加权重要性的矩阵。通过将该矩阵按位置注意力分数总和进行归一化处理,得到相对注意力谱。
下一步是将序列及其对应注意力谱转换为PSPM矩阵。首先需要在序列特定位置累加各氨基酸的注意力谱,通过遍历序列并确保每个序列长度与其关联的注意力谱相匹配来保持数据完整性。使用Logomaker Python语言包33生成蛋白质序列标识图,将PSPM作为Logomaker对象的输入。生成PSPM的具体算法详见补充图S42。
Mutation analysis
变异分析
Data for mutation analysis was compiled from dbSNP, which annotates pathogenic mutations occurring directly at or near modification sites. Uniprot identifiers for each gene were mapped using https://www. uniprot.org/id-mapping to extract the full-length protein sequences used in our analysis. Subsequently, wild-type and mutant protein sub sequences were prepared. For each wild-type sub sequence (with the modified amino acid residue at the center), 399 mutant sequences were generated. This was achieved by substituting each amino acid residue in the wild-type sequence with one of the 20 standard amino acids, excluding the original residue. Given the 21-length of the wildtype sub sequences, the total possible combinations to generate pointmutated sub sequences amounted to $21\times19$ . Our method produced all possible single amino acid substitutions for each wild-type sequence at every position. Inference prompts for both wild and mutant subsequences were then processed through the model to generate probability scores. These scores indicate whether any point mutations in the wildtype sub sequence affected the model’s output by altering PTM regulation. Initially, the probability score for the wild-type subsequence was obtained (the probability of the model generating a ‘POSITIVE’ token signifying modification). Subsequently, we analyzed the probability scores for all mutant sub sequences to determine whether specific point mutations caused a significant change in PTM regulation (probability of the model generating a’NEGATIVE’ token, signifying PTM down regulation). Heatmaps and line plots were utilized to analyze the average effect of mutations by position, based on the output probabilities generated by PTMGPT2.
用于突变分析的数据来自dbSNP,该数据库注释了直接发生在修饰位点或其附近的致病突变。每个基因的Uniprot标识符通过https://www.uniprot.org/id-mapping进行映射,以提取我们分析中使用的全长蛋白质序列。随后,制备了野生型和突变型蛋白质亚序列。对于每个野生型亚序列(以修饰氨基酸残基为中心),生成了399个突变序列。这是通过将野生型序列中的每个氨基酸残基替换为20种标准氨基酸之一(不包括原始残基)来实现的。鉴于野生型亚序列的长度为21,生成点突变亚序列的总可能组合为$21\times19$。我们的方法为每个野生型序列的每个位置生成了所有可能的单氨基酸替换。然后通过模型处理野生型和突变型亚序列的推理提示,以生成概率分数。这些分数表明野生型亚序列中的任何点突变是否通过改变PTM(翻译后修饰)调控影响了模型的输出。首先,获得野生型亚序列的概率分数(模型生成表示修饰的“POSITIVE”标记的概率)。随后,我们分析了所有突变亚序列的概率分数,以确定特定的点突变是否引起PTM调控的显著变化(模型生成表示PTM下调的“NEGATIVE”标记的概率)。基于PTMGPT2生成的输出概率,利用热图和线图按位置分析突变的平均效应。
Reporting summary
报告摘要
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
有关研究设计的更多信息可在本文链接的《Nature Portfolio报告摘要》中获取。
Data availability
数据可用性
Training and benchmark datasets for all 19 PTMs are publicly available at Zenodo: https://doi.org/10.5281/zenodo.11377398. Trained PTMGPT2 models are available at https://zenodo.org/records/ 11362322 and https://doi.org/10.5281/zenodo.11371883. Source data are provided with this paper.
全部19个PTM的训练和基准数据集已在Zenodo公开提供:https://doi.org/10.5281/zenodo.11377398。训练完成的PTMGPT2模型可通过https://zenodo.org/records/11362322 和 https://doi.org/10.5281/zenodo.11371883 获取。源数据随本文提供。
Code availability
代码可用性
The source code for PTMGPT2 is publicly accessible at https://github. com/pallucs/ PTMGPT2, and the repository includes files essential for conducting training and inference procedures. Model: This folder hosts a sample model designed to predict PTM sites from given protein sequences, illustrating PTMGPT2’s application. Tokenizer: This folder contains a sample tokenizer responsible for tokenizing protein sequences, including handcrafted tokens for specific amino acids or motifs. Inference.ipynb: This file provides executable code for applying PTMGPT2 model and tokenizer to predict PTM sites, serving as a practical guide for users to apply the model to their datasets.
PTMGPT2的源代码可在 https://github.com/pallucs/PTMGPT2 公开访问,该代码库包含进行训练和推理程序所必需的文件。
模型 (Model) : 该文件夹托管了一个样本模型,旨在从给定蛋白质序列预测PTM位点,展示了PTMGPT2的应用。
分词器 (Tokenizer) : 该文件夹包含一个样本分词器,负责对蛋白质序列进行分词处理,包括针对特定氨基酸或基序的手工设计token。
Inference.ipynb : 该文件提供了可执行代码,用于应用PTMGPT2模型和分词器来预测PTM位点,为用户将模型应用于自身数据集提供了实用指南。
References
参考文献
Acknowledgements
致谢
This work was supported in part by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1A2C2005612 to K.T.C.) and (No. 2022R1G1A1004613 to H.T.), in part by the Human Resources Program in Energy Technology of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) with financial resources from the Ministry of Trade, Industry & Energy, Republic of Korea (No. 20204010600470 to K.T.C.), and in part by the Korea Big Data Station (K-BDS) with computing resources and technical support.
本研究部分由韩国国家研究基金会 (NRF) 资助的韩国政府 (MSIT) 项目 (编号 2020R1A2C2005612 授予 K.T.C.) 和 (编号 2022R1G1A1004613 授予 H.T.) 支持,部分由韩国能源技术评估与规划研究院 (KETEP) 的能源技术人力资源项目 (编号 20204010600470 授予 K.T.C.) 提供资金支持,该项目资金来源于韩国贸易、工业和能源部,部分由韩国大数据站 (K-BDS) 提供计算资源和技术支持。
Author contributions
作者贡献
P.S., J.K., H.T., and K.T.C: conceptualization, methodology, investigation, writing review & editing and validation. P.S. and J.K.: writing original draft, Data Curation, implementation, software and website development, and visualization. H.T., and K.T.C: Supervision and project administration.
P.S., J.K., H.T. 和 K.T.C: 概念化、方法论、调研、文稿审阅与编辑及验证。P.S. 和 J.K.: 初稿撰写、数据管理、实施、软件与网站开发及可视化。H.T. 和 K.T.C: 监督与项目管理。
Competing interests
利益冲突
The authors declare no competing interests.
作者声明无竞争性利益。
Additional information Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41467-024-51071-9.
补充信息
在线版本包含补充材料,可在 https://doi.org/10.1038/s41467-024-51071-9 获取。
Correspondence and requests for materials should be addressed to Hilal Tayara or Kil To Chong.
材料相关问询及请求请致函 Hilal Tayara 或 Kil To Chong。
Peer review information Nature Communications thanks Dong Xu and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
同行评审信息:Nature Communications 感谢 Dong Xu 及其他匿名审稿人对本工作的同行评审所作出的贡献。同行评审文件可供查阅。
Reprints and permissions information is available at http://www.nature.com/reprints
可在 http://www.nature.com/reprints 获取转载与许可信息
Publisher’s note Springer Nature remains neutral with regard to juris diction al claims in published maps and institutional affiliations.
出版方注:施普林格·自然对出版地图和机构附属中的管辖权主张保持中立。
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit creative commons.org/licenses/by-nc-nd/4.0/.
开放获取 本文遵循知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International License) 。该协议允许在任何媒介或格式中进行非商业性使用、共享、分发和复制,但必须:适当标注原作者姓名及来源;提供知识共享许可协议的链接;并注明是否对授权材料进行了修改。本协议不允许分享基于本文或其部分内容改编的衍生材料。除非在材料标注中另有说明,本文中的图片或其他第三方材料均包含在文章的知识共享许可范围内。如果材料未包含在文章的知识共享许可中,且您的预期用途未被法律法规允许或超出许可范围,您需要直接向版权持有人获取授权。要查看该许可的副本,请访问 http://creativecommons.org/licenses/by-nc-nd/4.0/。
$\circledcirc$ The Author(s) 2024
$\circledcirc$ The Author(s) 2024